

Abstract 抽象的
Hundreds of microbiota gene expressions are significantly different between healthy and diseased humans. The “bottleneck” preventing a mechanistic dissection of how they affect host biology/disease is that many genes are encoded by nonmodel gut commensals and not genetically manipulatable. Approaches to efficiently identify their gene transfer methodologies and build their gene manipulation tools would enable mechanistic dissections of their impact on host physiology. This paper will introduce a step‐by‐step protocol to identify gene transfer conditions and build the gene manipulation tools for nonmodel gut microbes, focusing on Gram‐negative Bacteroidia and Gram‐positive Clostridia organisms. This protocol enables us to identify gene transfer methods and develop gene manipulation tools without prior knowledge of their genome sequences, by targeting bacterial 16s ribosomal RNAs or expanding their compatible replication origins combined with clustered regularly interspaced short palindromic repeats machinery. Such an efficient and generalizable approach will facilitate functional studies that causally connect gut microbiota genes to host diseases.
数百种微生物基因的表达在健康人和患病人群之间存在显著差异。阻碍我们对其如何影响宿主生物学/疾病进行机制解析的“瓶颈”在于,许多基因是由非模式肠道共生菌编码的,且无法进行基因操作。高效识别其基因转移方法并构建其基因操作工具的方法,将有助于我们对其对宿主生理学的影响进行机制解析。本文将介绍一种分步方案,用于识别基因转移条件并构建针对非模式肠道微生物的基因操作工具,重点关注革兰氏阴性拟杆菌和革兰氏阳性梭菌 。该方案使我们能够在不了解其基因组序列的情况下,通过靶向细菌 16s 核糖体 RNA 或扩展其相容的复制起点,并结合成簇的规律间隔短回文重复序列机制,来识别基因转移方法并开发基因操作工具。这种高效且可推广的方法将促进肠道微生物基因与宿主疾病因果关系的功能研究。
Keywords: genetic manipulation strategies, human gut microbiota, nonmodel gut Bacteroidia
, nonmodel gut Clostridia
关键词: 基因操作策略、人类肠道微生物群、非模型肠道拟杆菌
,非模型肠道梭菌
A protocol introducing a step‐by‐step genetic manipulation method would facilitate the investigation of those functional genes encoded by nonmodel gut commensals. The gene‐editing tools could be established in nonmodel gut Bacteroidia and Clostridia without prior knowledge of their genome information. The genetic manipulation pipeline may serve as a high‐throughput genetics screening and manipulating platform for human gut microbes.
引入分步遗传操作方法的方案将有助于研究非模式肠道共生菌编码的功能基因。该基因编辑工具可在非模式肠道拟杆菌和梭菌中建立,而无需事先了解其基因组信息。该遗传操作流程可作为人类肠道微生物的高通量遗传学筛选和操作平台。

Highlights 亮点
A protocol introducing a step‐by‐step genetic manipulation method would facilitate the investigation of those functional genes encoded by nonmodel gut commensals.
引入逐步基因操作方法的协议将有助于研究非模型肠道共生体编码的功能基因。The gene‐editing tools could be established in nonmodel gut Bacteroidia and Clostridia without prior knowledge of their genome information.
可以在非模型肠道拟杆菌和梭菌中建立基因编辑工具,而无需事先了解它们的基因组信息。The genetic manipulation pipeline may serve as a high‐throughput genetics screening and manipulating platform for human gut microbes.
基因操作流程可作为人类肠道微生物的高通量遗传学筛选和操作平台。
INTRODUCTION 介绍
The gut microbiota impacts human biology in many ways. Multiomics studies revealed many microbiota genes whose expressions significantly differ between healthy and diseased humans [1, 2, 3, 4, 5, 6]. However, unraveling the causal molecular mechanisms underlying microbiota gene–host biology interactions remains challenging, mainly due to limited approaches to precisely manipulating these disease/biology‐associated microbes and their metabolic genes.
肠道菌群以多种方式影响着人体生物学。多组学研究揭示,许多菌群基因的表达在健康人和患病人群中存在显著差异 [ 1 , 2 , 3 , 4 , 5 , 6 ]。然而,揭示菌群基因与宿主生物学相互作用背后的分子机制仍然充满挑战,这主要是因为目前精准操控这些疾病/生物学相关微生物及其代谢基因的方法有限。
Developing genetic manipulation tools for nonmodel gut microbes is necessary because: (1) Previous studies have revealed that host diseases are significantly associated with microbiota genes [1, 2, 3, 4, 5, 6]. Those genes are mostly expressed in nonmodel gut microbes that are not genetically tractable. Establishing genetic tools will be the first step to manipulating their gene expression within the host, and further to study their impact on human diseases. (2) Human biology is profoundly regulated by gut microbiota, yet the knowledge about which gut microbes and genes play an essential role remains largely unstudied. Genetic manipulation tools will facilitate the functional studies of physiology interactions between gut microbiota and host.
开发非模型肠道微生物的遗传操作工具十分必要,因为:(1) 先前的研究表明,宿主疾病与微生物基因密切相关 [ 1 , 2 , 3 , 4 , 5 , 6 ]。这些基因大多在非模型肠道微生物中表达,且不易被遗传调控。建立遗传工具将是操纵这些基因在宿主体内表达的第一步,并进一步研究它们对人类疾病的影响。(2) 人体生物学深受肠道微生物的调控,但关于哪些肠道微生物和基因发挥重要作用的知识仍未被深入研究。遗传操作工具将有助于肠道微生物与宿主之间生理相互作用的功能研究。
Here, we reported a step‐by‐step protocol to build the genetic manipulation strategies for nonmodel gut Bacteroidia and Clostridia microbes, whose abundances dominate healthy human guts [7, 8]. By targeting bacterial 16s ribosomal RNAs (rRNAs) or expanding their compatible replication origins combined with clustered regularly interspaced short palindromic repeats (CRISPR) machinery, this pipeline enables us to identify exogenous genomic DNA transfer methodologies and develop genetic tools without prior knowledge of the genome sequence of those nonmodel gut microbes.
GENETIC MANIPULATION STRATEGIES
Identifying gene transfer methods for Bacteroidia microbes and building their gene insertion tools
Escherichia coli (E. coli) conjugation was used to introduce the exogenous DNA into the recipient microbes, as the method has been proven effective in some Bacteroides and Clostridium [9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20]. Culture conditions of strains on agar plates and in liquid broth were first screened and then these microbes were tested against a collection of antibiotics to (1) find an antibiotic they are susceptible to, so its resistance gene will be used as a universal selective marker, and (2) identify an antibiotic the recipient microbe is resistant to but not the donor E. coli, so it can be used as an additive to suppress E. coli growth after conjugation.
The Bacteroidia are Gram‐negative obligate anaerobes that uptake exogenous DNA and have efficient homologous recombination (HR) [16]. To establish a generalizable approach for the genetic manipulation of Bacteroidia (Bacteroides and Prevotella) microbes, conserved bacterial 16s rRNA gene, whose sequence has been widely used to assess microbiome diversity and construct bacterial phylogeny, was selected as a universal target. To do this, a synthesized chimeric 16s (chi‐16s) sequence, with high homology to the Bacteroidia 16s rRNA genes, was assembled with a suicide conjugation vector, and transported into recipient Bacteroidia microbes. Those transconjugants whose 16s rRNA loci have been inserted by the suicide vector will be genetically tractable.
Materials and devices
Primer star DNA polymerase (Takara, Cat# R045), Blue sapphire DNA polymerase (Takara, Cat# RR350), Plasmid Midiprep Kit (Zymo Research, Cat# D4201), DNA Clean and Concentrator (Zymo Research, Cat# D4003), Tryptic Soy Agar (BD, Cat# 236950), Brain Heart Infusion Agar (BD, Cat# 241830), Columbia Blood Agar (CBA) (BD, Cat# 279240), Horse blood (Hemostat Laboratories, Cat# 637291), Luria–Bertani (LB) broth (BD, Cat# BP1426), glycerol (Fisher Bioreagents, Cat# BP229), the vacuum‐pumping system, phosphate‐buffered saline (PBS) (Gibco, Cat# 10010‐031), centrifuge, polymerase chain reaction (PCR) amplifier, d‐cycloserine (D) (TCI, Cat# C1189), gentamicin (G) (GoldBio, Cat# G‐400‐25), kanamycin (K) (GoldBio, Cat# K‐120‐25), carbenicillin (GoldBio, Cat# C‐103‐25), thiamphenicol (Thiam) (Acros Organics, Cat# 455450250), anaerobic chamber, aerobic incubator, electroporation system, Thermo Scientific Nanodrop 2000, Gibson Assembly Cloning Kit (NEB, Cat# E5510S), Quick DNA fungal/bacterial kit (Zymo Research, Cat# D6005), 50 mL Tube Top Vacuum Filter System (0.22 mm) (Corning Life Sciences, Cat. #430320), and ultralow temperature freezer.
Screening of culture conditions
The culture of Gram‐negative Bacteroidia strains was incubated in an anaerobic chamber at 37°C under an atmosphere of 5% carbon dioxide (CO2), 7.5% hydrogen (H2), and 87.5% nitrogen (N2). The agar plates were left in the anaerobic chamber for at least one overnight before use. The liquid medium was left in the chamber with a loosened cap for at least 48 h before inoculation.
Strains were restreaked (from original glycerol stock) onto pre‐reduced agar plates (such as Tryptic Soy Agar + 5% blood [TSAB] plates, Brain Heart Infusion Agar + 5% blood [BHIB] plates, or CBA plates). Then, those that can grow on agar plates were subcultured into pre‐reduced liquid medium: Mega, Chopped Meat Medium (CMM), and Reinforced Clostridial Medium (RCM, BD, Cat# 218081). Strains that can grow in any of the four liquid cultures were subjected to the antibiotics test (Figure 1A).
Figure 1.

Workflow for identifying gene transfer methods for Bacteroidia microbes and developing genetic manipulation tools for Bacteroidia microbes via single crossover insertion and double crossover deletion. (A) Workflow for identifying gene transfer methods for Bacteroidia microbes. (B, C) Genetic manipulation of asparaginase gene (ansB) in Prevotella bivia DSM 20514 via single crossover integration. (B) Diagnostic polymerase chain reaction (PCR) showed that the mutant strain (ΔansB) had the PCR product of ~2 kb, while the wide type (WT) strain had no band. (C) Liquid chromatography–mass spectrometry trace showed that the mutant strain (ΔansB) lost the ability to convert substrate asparagine to aspartic acid. (D, E) Genetic manipulation of thymidine kinase gene (tdk) in Bacteroides sp. 1_1_30 via double crossover deletion. (D) Schematic view of knocking out gene tdk in gut bacteria Bacteroides sp. 1_1_30 using a double crossover recyclable marker system. (E) Diagnostic PCR showed that the mutant strain (Δtdk) had the expected shorter PCR product compared with the WT strain. BHIB, Brain Heart Infusion Agar + Horse blood; CBA, Columbia Blood Agar; chi‐16s, chimeric 16s; CMM, Chopped Meat Medium; D, d‐cycloserine; diagF, diagnostic forward primer; diagR, diagnostic reverse primer; E. coli, Escherichia coli; G, gentamicin; LB, Luria–Bertani; RCM, Reinforced Clostridial Medium; rRNA, ribosomal RNA; seqR, sequencing primer (reverse); Thiam, thiamphenicol; TSAB, Tryptic Soy Agar + Horse blood.
Keynotes: For the screening of the culture conditions of liquid medium, Bacteroidia strains need to be first recovered on agar plates to ensure Bacteroidia strains are activated, instead of inoculating Bacteroidia strains into liquid medium from the frozen glycerol stock directly.
Potential issues and solutions: If the Bacteroidia strains of interest cannot grow on the common agar plates or in the liquid medium listed above, other specific plates or liquid mediums that favor the growth of the target strains need to be used to screen the culture conditions.
Antibiotic test
To find the antibiotic that suppresses the growth of conjugation donor E. coli S17, the Bacteroidia (Bacteroides and Prevotella) microbes were restreaked on agar plates supplemented with 200 µg/mL gentamicin (G) or 250 µg/mL d‐cycloserine (D). The tested Bacteroidia microbes are expected to grow on plates with either gentamicin or d‐cycloserine. Most of the Bacteroidia strains we tested so far are sensitive to thiamphenicol (Thiam), so the thiamphenicol‐resistant gene (catP) can be used as a universal marker to select transconjugants whose genome was integrated by the suicide vector. The minimum inhibitory concentrations (MICs) of thiamphenicol of the Bacteroidia microbes were tested on agar plates containing thiamphenicol at different concentrations (Figure 1A).
Keynotes: Antibiotics need to be added into the agar medium before the agar plates are poured and solidified; agar plates supplemented with antibiotics only on the surface of the plates may cause misleading antibiotic test results.
Potential issues and solutions: If the Bacteroidia strains of interest are not resistant against gentamicin or d‐cycloserine, other specific antibiotics that can suppress the growth of E. coli S17 can be used for the test. Likewise, if the Bacteroidia strains of interest are resistant against thiamphenicol, the antibiotic marker on the suicide vector can be replaced by other markers, whose corresponding antibiotics can suppress the growth of the Bacteroidia strains.
Vector assembly
By assembling the synthesized conserved ~1 kb chi‐16s rRNA sequence (chi‐16s) for Bacteroidia with the suicide vector pExchange [21], plasmids pGM‐NACB (targeting 16s rRNA gene of Bacteroides, GM: genetic manipulation, N: no gram‐positive replication origin, A: R6K gram‐negative replication origin, C: catP antibiotic marker, B: Bacteroides) and pGM‐NACP (targeting 16s rRNA gene of Prevotella, P: Prevotella) were generated [2] (Figure 1A). Likewise, to target other specific genes in Bacteroidia strains, an ~1 kb fragment of the target gene could be amplified by PCR and assembled with the backbone amplified from the suicide vector pGM‐NACB/P to get the plasmid for mutating the target gene via insertion–deletion (Figure 1A).
Keynotes: In the case the size of the target gene is smaller than 1 kb, a ~0.5 kb fragment of the target gene is also functional for the single crossover, the key is that the fragment has to be an incomplete fraction (both upstream and downstream) of the target gene.
Introduction of suicide vectors into the Bacteroidia microbes
The suicide vectors pGM‐NACB/P were introduced into the target Bacteroides/Prevotella using E. coli conjugation following the previously published protocol [21]. A single colony of the target Bacteroidia microbe was inoculated in 3 mL liquid broth and cultured in an anaerobic chamber at 37°C. The E. coli S17 harboring the pGM‐NACB/P vector was inoculated in the LB broth supplemented with carbenicillin (100 µg/mL) and grown at 37°C with aerobic shaking at 220 rpm. After ~12–16 h, when the OD600 of E. coli S17 reached 0.8–1.0, 6 mL of E. coli S17 culture was centrifuged at 1500g for 2 min. The supernatant was discarded, and the cell pellet was washed twice with 3 mL PBS buffer (pH = 7.4). The washed E. coli S17 cell pellet was resuspended in a 3 mL overnight culture of the target Bacteroidia strain and gently mixed by pipetting. The mixture was filtered through a 0.2 µm filter. The filtered liquid was discarded. The filter with the mixture of donor and recipient cells was placed onto the surface of a pre‐reduced agar plate (cell surface facing down). The plate was incubated aerobically in a 37°C incubator.
After incubation aerobically at 37°C for 24 h, the filter was soaked in 2 mL pre‐reduced liquid broth. The cell on the filter was resuspended into the broth by gentle vortexing. The mixture was then transferred into the anaerobic chamber, and 100 µL (or serial diluted suspension) was plated onto a pre‐reduced agar plate with 200 µg/mL gentamicin + 15 µg/mL thiamphenicol (or MICs). Colonies of the target strain typically appeared after 36–48 h. Four colonies were picked and restreaked on a pre‐reduced agar plate with 200 µg/mL gentamicin + 15 µg/mL thiamphenicol (or MICs) to isolate single colonies.
Keynotes: The target Bacteroidia microbes are cultured in the anaerobic chamber overnight (~12 h). Do not culture the Bacteroidia strains for too long before conjugation, it will lead to the lysis of the strains.
Potential issues and solutions: For some Bacteroidia strains that are pretty sensitive to oxygen, the aerobic conjugation will lead to the death of the majority of bacteria, in this case, there are two solutions: (1) expend the Bacteroidia strains by recovering the bacteria on the filter in liquid medium (supplemented with antibiotics to suppress the growth of E. coli) in the anaerobic chamber, then plate the growth in the liquid medium onto plates with antibiotics to isolate transconjugants; and (2) perform the conjugation on the filter in the anaerobic chamber.
Diagnostic PCR and sequencing to verify the single crossover integration
The isolated single colony was inoculated in a 3 mL liquid broth supplemented with 200 µg/mL gentamicin + 15 µg/mL thiamphenicol (or MICs). After 12 h, genomic DNA was extracted using a Quick DNA fungal/bacterial kit (Zymo Research). Diagnostic PCR was performed using primers 16s_27F and R6K_R to verify the single crossover integration of pGM‐NACB/P at their 16s rRNA loci. An ~2.5 kb PCR band would be seen in the transconjugants, whose chromosomal 16s rRNA loci were integrated by pGM‐NACB/P. The 2.5 kb PCR product was purified using a DNA Clean & Concentrator kit (Zymo Research) and sent for sequencing using primer R6K_F_RC. The sequencing results would show that a partial sequence of the 2.5 kb fragment came from the synthetic chi‐16s in pGM‐NACB/P and a partial sequence of the original 16s rRNA gene of the target strain, suggesting a single crossover of pGM‐NACB/P into one of its 16s rRNA loci (Figure 1A).
This single crossover integration strategy readily applies to other genes of interest in Bacteroidia microbes. To inactive a target gene in Bacteroidia strains, ~1 kb fragment of the target gene is amplified and the purified PCR product is then Gibson‐assembled, with the backbone amplified from the suicide vector pGM‐NACB, to get the plasmid for the target gene (Figure 1A). The plasmid is transferred into the conjugation donor E. coli S17 and introduced into the recipient microbe via conjugation. The transconjugants that undergo the expected single crossover integration are identified by diagnostic PCR and sequencing.
Leveraging this protocol, after using the 16s rRNA‐targeting strategy to identify the gene transfer method for a nonmodel gut microbe Prevotella bivia DSM 20514, we were able to inactivate the asparaginase gene (ansB, which catalyzes the conversion of asparagine to aspartic acid) via single crossover insertion, as shown in Figure 1B,C. In diagnostic PCR, the mutant strain (ΔansB) had the PCR product of ~2 kb, while the wide type (WT) strain had no band (Figure 1B), and we also demonstrated that the mutant strain lost the ability to convert substrate asparagine to aspartic acid by liquid chromatography–mass spectrometry (LC‐MS) (Figure 1C).
On the basis of the essential first step that assesses the tractability of nonmodel Bacteroidia microbes, including Prevotella, our approach also paves the way for developing more advanced genetic tools, such as the double crossover recyclable marker system. As a proof of concept, after the establishment of the 16s rRNA‐targeting strategy in gut bacteria Bacteroides sp. 1_1_30, we further developed the double crossover recyclable marker system to knock out the thymidine kinase gene (tdk, which phosphorylates both thymidine and deoxyuridine). As shown in Figure 1D,E, following the integration of the suicide plasmid guided by the left arm and right arm of the targeted gene via HR, the antibiotic marker catP will be removed in the second step (double crossover) to get the markerless mutant strain (Figure 1D), in diagnostic PCR, the mutant strain (Δtdk) had the expected shorter PCR product compared with the WT strain (Figure 1E).
Potential issues and solutions: If there is no correct transconjugant out of the four restreaked colonies upon diagnostic PCR, suggesting that the integration efficiency is low, in this case, solution 1 is to pick more colonies (like 24 colonies) to restreak and then do diagnostic PCR, solution 2 is to amplify another version of a fragment from the target gene.
Experimental results interpretation: For the result of diagnostic PCR of the transconjugants, as shown in Figure 1A,B, because the forward diagnostic primer binds the sequence on the genome and the reverse diagnostic primer binds the sequence on the plasmid, only transconjugants that undergo the expected integration would have the 2.5 kb PCR product; WT strain or transconjugants that undergo the unexpected insertion would not have the 2.5 kb PCR product.
Identifying methods for Clostridia microbes to uptake and stably maintain exogenous genomic DNA
For nonmodel Clostridia microbes, culture conditions on agar plates and in liquid broth were screened and identified. For antibiotic resistance, a collection of antibiotics was also screened using a method similar to that used in Bacteroidia strains. Likewise, E. coli conjugation was used to transport exogenous DNA into the recipient Clostridia microbes.
Compared with the Bacteroidia strains, the Gram‐positive Clostridia gut microbes are more resistant to genetic manipulations for two reasons: (1) It is challenging to deliver exogenous DNA to Clostridia strains. E. coli conjugation is commonly used to transfer a plasmid with a compatible replication origin (rep ori) to a recipient Clostridia microbe. The rep ori allows the recipient to stably maintain exogenous DNA within the bacteria. (2) Clostridia microbes have very inefficient HR.
Previous studies have used the Group II intron (ClosTron) to introduce genome insertion [15] or the CRISPR‐Cas9 to induce chromosomal double‐strand break to promote the selection of HR [10, 11]. Both genetic components need to be assembled with a compatible rep ori to be maintained stably in the recipient strain. Therefore, identifying a Clostridia‐compatible rep ori will be the first step toward developing the genetic tools for Clostridia microbes. Their rep oris were expanded (from 4 to 9) to identify a compatible rep ori for nonmodel gut Clostridia strains [22, 23, 24]. A mixed‐conjugation strategy was developed to identify exogenous gene transfer methods for Clostridia strains on a large scale.
Materials and devices
Primer star DNA polymerase (Takara, Cat# R045), Blue sapphire DNA polymerase (Takara, Cat# RR350), Plasmid Midiprep Kit (Zymo Research, Cat# D4201), DNA Clean and Concentrator (Zymo Research, Cat# D4003), Tryptic Soy Agar (BD, Cat# 236950), Brain Heart Infusion Agar (BD, Cat# 241830), CBA (BD, Cat# 279240), Horse blood (Hemostat Laboratories, Cat# 637291), LB broth (BD, Cat# BP1426), glycerol (Fisher Bioreagents, Cat# BP229), PBS (Gibco, Cat# 10010‐031), centrifuge, PCR amplifier, tetracycline (GoldBio, Cat# T‐101‐25), chloramphenicol (VWR, Cat# 0230), d‐cycloserine (D) (TCI, Cat# C1189), gentamicin (G) (GoldBio, Cat# G‐400‐25), kanamycin (K) (GoldBio, Cat# K‐120‐25), carbenicillin (GoldBio, Cat# C‐103‐25), thiamphenicol (Thiam) (Acros Organics, Cat# 455450250), anaerobic chamber, aerobic incubator, electroporation system, Thermo Scientific Nanodrop 2000, Gibson Assembly Cloning Kit (NEB, Cat# E5510S), Quick DNA fungal/bacterial kit (Zymo Research, Cat# D6005), and ultralow temperature freezer.
Screening of culture conditions
The culture conditions for the Gram‐positive Clostridia microbes were screened. Strains were restreaked (from the original glycerol stock) onto pre‐reduced agar plates (such as TSAB, BHIB, or CBA plates). Then, microbes that can grow on these agar plates were subcultured into 1 mL pre‐reduced liquid medium: Mega, CMM, and RCM. Strains that can grow in any one of the four liquid cultures were subject to the antibiotics test (Figure 2).
Figure 2.
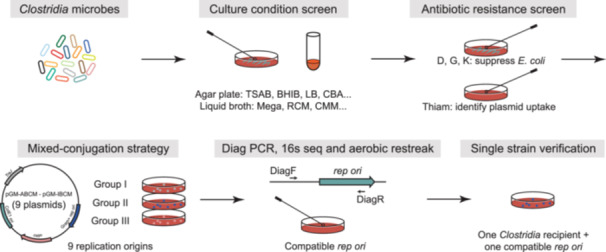
Workflow for the identification of methods for Clostridia microbes to uptake and stably maintain exogenous genomic DNA. BHIB, Brain Heart Infusion Agar + Horse blood; CBA, Columbia Blood Agar; CMM, Chopped Meat Medium; D, d‐cycloserine; E. coli, Escherichia coli; G, gentamicin; K, kanamycin; LB, Luria–Bertani; PCR, polymerase chain reaction; RCM, Reinforced Clostridial Medium; rep ori, replication origin; TSAB, Tryptic Soy Agar + Horse blood.
Keynotes: For the screening of the culture conditions of liquid medium, Clostridia strains need to be first recovered on agar plates to ensure that Clostridia strains are activated, instead of inoculating Clostridia strains into the liquid medium from the frozen glycerol stock directly.
Potential issues and solutions: If the Clostridia strains of interest cannot grow on the common agar plates or in the liquid medium listed above, other specific plates or liquid mediums that favor the growth of the target strains need to be used to screen the culture conditions.
Antibiotic test
The Clostridia strains were restreaked on agar plates supplemented with 250 µg/mL d‐cycloserine (D), 200 µg/mL gentamicin (G), or 200 µg/mL kanamycin (K). d‐cycloserine or gentamicin will be used to inhibit the growth of conjugation donor E. coli CA434 after conjugation, and kanamycin will be used to inhibit the growth of conjugation donor E. coli HB101/pRK24. Both E. coli CA434 and HB101/pRK24 have been shown to successfully conjugate exogenous genomic DNA into Clostridium bacteria like Clostridium sporogenes or Clostridium acetobutylicum in previous studies [10, 11, 15].
The majority of the Clostridia strains we tested so far are sensitive to thiamphenicol, so the thiamphenicol‐resistant gene (catP) can be exploited as a universal marker to select transconjugants that can uptake and maintain exogenous genomic DNA. The MICs of thiamphenicol of the Clostridia microbes were tested on agar plates containing thiamphenicol at different concentrations (Figure 2).
Keynotes: Antibiotics need to be added into the agar medium before the agar plates are poured and solidified; agar plates supplemented with antibiotics only on the surface of the plates may cause misleading antibiotic test results.
Potential issues and solutions: If the Clostridia strains of interest are not resistant against d‐cycloserine, gentamicin, or kanamycin, other specific antibiotics that can suppress the growth of E. coli CA434 or HB101/pRK24 can be used for the test. Likewise, if the Clostridia strains of interest are resistant against thiamphenicol, the antibiotic marker on the conjugation vector can be replaced by other markers, whose corresponding antibiotics can suppress the growth of the Clostridia strains.
Vector assembly
A series of vectors pGM‐xBCM (pGM‐ABCM, BBCM, CBCM, DBCM, EBCM, FBCM, GBCM, HBCM, and IBCM) harboring different rep oris for Clostridia microbes were generated to screen the compatible rep oris for the recipient Clostridia strains [2]. To include more rep oris for screening, the rep ori sequences could be amplified by PCR and assembled with the backbone amplified from pGM‐xBCM.
Mixed‐conjugation strategy to identify Clostridia microbes that uptake and maintain exogenous plasmid DNA
The series of vectors pGM‐ABCM, BBCM, CBCM, DBCM, EBCM, FBCM, GBCM, HBCM, and IBCM harboring different rep oris, but the same antibiotic marker catP (against thiamphenicol) were transformed into chemical competent E. coli CA434 or E. coli HB101/pRK24. Mixed‐conjugation strategies separating these E. coli donors into three groups (Group I: pGM‐ABCM, BBCM, and CBCM; Group II: pGM‐DBCM, EBCM, and FBCM; and Group III: pGM‐GBCM, HBCM, IBCM) were established to identify the compatible rep ori for each Clostridia microbe of interest. For the Clostridia microbes resistant to d‐cycloserine (250 μg/mL) or gentamicin (200 µg/mL), E. coli CA434 would be used as the conjugation donor. For the microbes that are not resistant to d‐cycloserine (250 μg/mL) or gentamicin (200 µg/mL) but resistant to kanamycin (200 μg/mL), E. coli HB101/pRK24 would be used as their conjugation donors.
The Clostridia microbe was restreaked on a pre‐reduced agar plate. After 24–48 h, a single colony was inoculated in 1 mL of liquid broth that supported its growth in an anaerobic chamber. On the same day, E. coli containing plasmids with different rep oris were inoculated into 6 mL of LB supplemented with tetracycline (15 µg/mL) and chloramphenicol (25 µg/mL) and shaken aerobically at 37°C for 12–18 h (overnight). The next day, these E. coli donors were separated into three groups, as mentioned above. For conjugating one Clostridia microbe, a 1.0 mL culture of each E. coli within the same group was mixed and centrifuged at 1500g for 2 min. The culture supernatant was discarded, and the cell pellet was gently washed with 500 µL PBS buffer (pH = 7.4). The PBS supernatant was then removed after centrifugation at 1500g for 2 min, and the cell pellet was transferred on ice into the anaerobic chamber. Next, the cell pellet (a total of three cell pellets) was mixed gently with 300 μL overnight culture of the targeting Clostridia microbe, and a 35 μL cell mixture was dotted on pre‐reduced agar plates. After 48 h, the cell dots were scraped using a sterile inoculation loop and resuspended in 300 μL pre‐reduced PBS (pH = 7.4) buffer. The cell suspension (100 µL) was plated on agar plates supplemented with 15 µg/mL thiamphenicol (or MICs) and 250 µg/mL d‐cycloserine or 200 µg/mL gentamicin (if E. coli CA434 is the conjugation donor), or 200 µg/mL kanamycin (if E. coli HB101/pRK24 is the conjugation donor). Colonies typically appeared after 36–48 h. Four colonies were picked and restreaked onto agar plates with the same antibiotics to isolate single colonies (Figure 2).
Keynotes: (1) The target Clostridia microbes are cultured in the anaerobic chamber overnight (~12 h), do not culture the Clostridia strains for too long before conjugation, which will lead to the lysis of the strains; (2) air dry the agar plates for conjugation a little bit is good for conjugation, if the plates are wet, the cell mixture dot will spread all over the plate, in this case, the cell mixture is diluted on the plate, which will reduce the conjugation efficiency.
Potential issues and solutions: In our experience, conjugation donor E. coli CA434 works better than E. coli HB101/pRK24. For specific strains of interest, if the two E. coli donors cannot transfer plasmids into the recipient strains, other E. coli donors could be utilized.
Diagnostic PCR and sequencing to verify the plasmid uptake
The isolated single colony was cultivated in 3 mL liquid broth supplemented with the corresponding antibiotics 250 µg/mL d‐cycloserine (or 200 µg/mL gentamicin/kanamycin) + 15 µg/mL thiamphenicol (or MICs). The genomic DNA was isolated from the resulting cell material using the Quick DNA fungal/bacterial kit (Zymo Research). Then multiplex diagnostic PCRs were performed to assess which plasmid was uptaken by the conjugation recipient Clostridia microbe. For the mixed‐conjugation with Group I (Groups II and III are performed likewise), primers pMTL_laz_diag_F (universal forward primer) + pGM‐ABCM_rep_R_1500bp + pGM‐BBCM_rep_R_1000bp + pGM‐CBCM_rep_R_2000bp (for 15 µL PCR reaction, the amount of the four primers is 0.75, 0.3, 0.3, and 0.3 µL [10 µM]) were used for diagnostic PCR. A PCR band of 1.5 kb (or 1.0 or 2.0 kb) would be seen if pGM‐ABCM (or BBCM or CBCM) is uptaken by the Clostridia microbe. In the meantime, to confirm that the picked and restreaked colonies are the target Clostridia strain but not the E. coli conjugation donor, the 16s rRNA region of the colony was amplified using primers 16s_27F + 16s_1391R. The PCR product was purified and sent for Sanger sequencing using primer 16s_1391R, and the colonies were further restreaked aerobically to confirm not to be E. coli (if the colonies cannot grow aerobically, they will be considered not to be E. coli) (Figure 2).
Keynotes: When performing the diagnostic PCR to figure out which plasmid in a group was transferred into the recipient strain, to avoid a false‐positive conclusion, it is necessary to include the genome of the recipient strain to serve as the negative control.
Experimental results interpretation: For the diagnostic PCR of each group (I, II, and III), when different plasmids are transferred into recipient strains, there will be PCR products of different sizes (1, 1.5, and 2 kb), the corresponding size of each plasmid in each group is annotated at the end of the “Name of primers” in Table S1.
Single E. coli donor‐conjugation validation
Next, single E. coli donor‐conjugation (one E. coli donor to one Clostridia recipient) was performed to validate that the PCR‐identified plasmid(s) can be conjugated into the targeted Clostridia microbe. A single colony of the targeted Clostridia strain was inoculated in a 1 mL liquid broth in an anaerobic chamber. The conjugation donor E. coli (CA434 or HB101/pRK24) harboring the PCR‐identified plasmid was inoculated into 6 mL of LB supplemented with tetracycline (15 µg/mL) and chloramphenicol (25 µg/mL) and shaken aerobically at 37°C for 12–18 h (overnight). After 12–18 h, 1.5 mL of the E. coli culture was centrifuged at 1500g for 2 min. The supernatant was discarded and the cell pellet was washed with 500 µL PBS buffer (pH = 7.4). The PBS supernatant was then removed after centrifugation at 1500g for 2 min, and the cell pellet was transferred on ice into the anaerobic chamber. Next, the cell pellet was mixed gently with a 300 μL overnight culture of the targeting Clostridia microbe, and a 35 μL cell mixture was dotted on pre‐reduced agar plates. After 48 h, the cell dots were scraped using a sterile inoculation loop and resuspended in 300 μL pre‐reduced PBS (pH = 7.4) buffer. The cell suspension (100 µL) was plated on agar plates supplemented with 15 µg/mL thiamphenicol (or MICs) and 250 µg/mL d‐cycloserine or 200 µg/mL gentamicin (if E. coli CA434 is the conjugation donor), or 200 µg/mL kanamycin (if E. coli HB101/pRK24 is the conjugation donor). Colonies typically appeared after 36–48 h. Four colonies were picked and restreaked onto agar plates with the same antibiotics to isolate single colonies. The isolated single colonies will be cultured in 1 mL of pre‐reduced liquid broth with the same antibiotics, and the glycerol stock will be prepared using the culture (Figure 2).
Keynotes: For the step of scraping cell dots and suspending cells in PBS to be plated onto agar plates with selective antibiotics, in the case that a lot of conjugations plates need to be scraped, it is better to scrap at most three plates and suspend in PBS at one time, keeping cells in PBS for too long will reduce the conjugation efficiency.
Potential issues and solutions: In the step of plating conjugation cells onto agar plates with d‐cycloserine (or gentamicin, kanamycin) + thiamphenicol (MICs), some recipient strains may overgrow because the MICs of thiamphenicol are not enough to suppress the growth of the plated cells that contain a high concentration of recipient strains; in this case, plates with a higher concentration of thiamphenicol need to be used (e.g., if the MICs of thiamphenicol are 7.5 µg/mL, 10 µg/mL thiamphenicol can be added into the agar plate).
Developing a CRISPRi‐dCpf1 lacZα system for Clostridia microbes
After identifying exogenous plasmid transfer methods for Clostridia strains, the next step is developing a tractable genetic tool that would function in those Clostridia commensals. Like Cas9‐mediated cutting and dCas9‐induced interference, CRISPR‐based genome editing systems have been recently used to manipulate C. sporogenes [10, 11] and Clostridium difficile [25]. In general, Clostridia has very inefficient HR, and the DNA double‐stranded break initiated by Cas9 or the like is mostly lethal. While much effort was spent finetuning a spectrum of conjugation parameters to identify the optimal condition for the Cas9 machinery in C. sporogenes, this condition is usually not readily applicable to other Clostridia commensals.
CRISPR interference deactivated Cpf1 (CRISPRi‐dCpf1) [26, 27, 28, 29, 30, 31] system was prioritized for Clostridia microbes because the dCpf1 does not initiate the DNA double‐strand break and is supposedly less toxic and applicable to a broader range of Clostridia compared with the Cas9/Cpf1. Indeed, plasmids carrying dCpf1 showed less toxicity and relatively higher conjugation efficiency than those with Cas9 or Cpf1. Combined with the CRISPRi‐dCpf1 machinery, lacZα was used as a transcription reporter to develop CRISPRi‐dCpf1 gene repression tools for nonmodel Clostridia microbes without prior knowledge of the genome sequence.
Materials and devices
Primer star DNA polymerase (Takara, Cat# R045), Blue sapphire DNA polymerase (Takara, Cat# RR350), Plasmid Midiprep Kit (Zymo Research, Cat# D4201), DNA Clean and Concentrator (Zymo Research, Cat# D4003), Tryptic Soy Agar (BD, Cat# 236950), Brain Heart Infusion Agar (BD, Cat# 241830), CBA (BD, Cat# 279240), Horse blood (Hemostat Laboratories, Cat# 637291), LB broth (BD, Cat# BP1426), glycerol (Fisher Bioreagents, Cat# BP229), PBS (Gibco, Cat# 10010‐031), centrifuge, PCR amplifier, tetracycline (GoldBio, Cat# T‐101‐25), chloramphenicol (VWR, Cat# 0230), d‐cycloserine (D) (TCI, Cat# C1189), gentamicin (G) (GoldBio, Cat# G‐400‐25), kanamycin (K) (GoldBio, Cat# K‐120‐25), thiamphenicol (Thiam) (Acros Organics, Cat# 455450250), anaerobic chamber, aerobic incubator, electroporation system, Thermo Scientific Nanodrop 2000, Gibson Assembly Cloning Kit (NEB, Cat# E5510S), Quick DNA fungal/bacterial kit (Zymo Research, Cat# D6005), ultralow temperature freezer, Direct‐zol RNA Microprep kit (Zymo Research, Cat# R2062), PrimeScript RT Reagent Kit (Takara, Cat# RR047A), real‐time quantitative PCR system (Applied Biosystems, ABI 7500).
Vector assembly
Three sets of plasmids pGM‐xBCD (pGM‐ABCD, BBCD, pGM‐CBCD, DBCD, EBCD, FBCD, GBCD, HBCD, and IBCD) carrying the CRISPRi‐dCpf1 machinery, plasmids pGM‐xBCL (pGM‐ABCL, BBCL, pGM‐CBCL, DBCL, EBCL, FBCL, GBCL, HBCL, and IBCL) carrying CRISPRi‐dCpf1 and the lacZα reporter gene, and plasmids pGM‐xBCF (pGM‐ABCF, BBCF, pGM‐CBCF, DBCF, EBCF, FBCF, GBCF, HBCF, and IBCF) carrying CRISPRi‐dCpf1, lacZα, and the lacZα targeting guide RNA (gRNA) were generated to test the CRISPRi‐dCpf1 lacZα system for Clostridia microbes [2] (Figure 3A–D). To target other specific genes in Clostridia strains, the gRNA locus targeting the promoter region and coding sequence (CDS) of the target gene (as in Figure 3D) could be amplified by PCR and assembled with the backbone amplified from pGM‐xBCD to get the plasmid carrying CRISPRi‐dCpf1 and the gRNA for the target gene.
Figure 3.
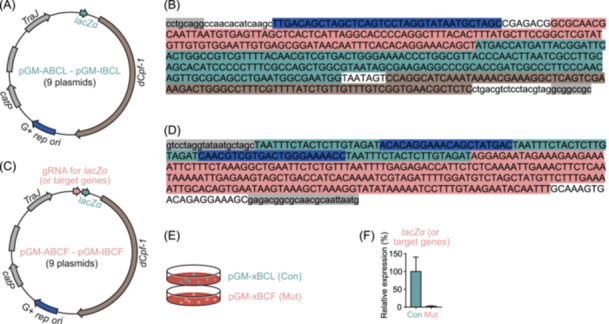
Development of a CRISPRi‐dCpf1 lacZα genetic manipulation system for Clostridia microbes. (A) Schematics of the set of plasmids pGM‐ABCL–pGM‐IBCL that carry the clustered regularly interspaced short palindromic repeats interference deactivated Cpf1 (CRISPRi‐dCpf1) machinery and the lacZα reporter gene. G+ rep ori, Gram‐positive replication origin. (B) The sequence of the lacZα locus consisting of guide RNA (gRNA) promoter PJ23119 (highlighted in blue), the lacZα promoter (in red), the lacZα coding sequence (in green), and lacZα terminator (in brown). The sequences highlighted in gray are restriction sites of SbfI and NotI, respectively. (C) Schematics of the set of plasmids pGM‐ABCF–pGM‐IBCF that carry the CRISPRi‐dCpf1 machinery, the lacZα reporter gene, and gRNA locus targeting the promoter region and coding sequence (CDS) of lacZα. (D) The sequence of the gRNA locus consisting of three dCpf1 direct repeat sequences (highlighted in green), two gRNA targeting both the promoter region and the template strand of lacZα (in blue), and terminator region obtained from the 16s rRNA gene of Clostridium sporogenes ATCC 15579 (CLOSPO_00916) (in red). The sequences highlighted in gray are homologous to the sequence in pGM‐ABCL. (E, F) Plasmids with compatible replication origins in the set of plasmids pGM‐ABCL–pGM‐IBCL (pGM‐xBCL, Control group, Con) and pGM‐ABCF–pGM‐IBCF (pGM‐xBCF, Mutant group, Mut) were introduced into the recipient Clostridia microbes, and the expression levels of lacZα were quantified by quantitative polymerase chain reaction (qPCR).
Utilization of dCpf1 to suppress the lacZα transcription in Clostridia strains
Using a Gram‐positive strain Clostridium bolteae DSM 29485 as an example, pGM‐ABCL and pGM‐ABCF were transformed into chemically competent E. coli CA434, respectively. E. coli CA434 harboring pGM‐ABCL and pGM‐ABCF were conjugated to Clostridium bolteae DSM 29485 (Figure 3E). The transconjugants were picked and restreaked onto a TSAB agar plate supplemented with d‐cycloserine (250 µg/mL) + thiamphenicol (15 µg/mL). Then, three isolated single colonies were cultivated in 5 mL Mega liquid broth supplemented with 15 µg/mL thiamphenicol for 36 h. The bacterial RNA was extracted using a Direct‐zol RNA Microprep kit (Zymo Research) and quantitative polymerase chain reaction (qPCR) was performed to quantify the relative expression of lacZα after normalizing to 16s rRNA gene, using primers dCpf1‐lacZα_qPCR_F and dCpf1‐lacZα_qPCR_R for lacZα gene and S74_16 s_qPCR_F and S74_16s_qPCR_R for the control 16s rRNA (Figure 3F).
This CRISPRi‐dCpf1 gene repression tool is readily applicable to other genes of interest in Clostridia microbes. To knock down a target gene in Clostridia strains, the gRNA locus targeting the promoter region and CDS of the target gene is introduced into the plasmid harboring dCpf‐1 and the corresponding compatible rep ori (the set of vectors pGM‐xBCD). As in targeting lacZα, the synthetic fragment containing the terminator region (Figure 3D, in red) was amplified to get a PCR product that has one direct repeat sequence (Figure 3D, in green) and gRNA (Figure 3D, in blue) fused with the terminator. Then, this PCR product was purified and used as the template for the second PCR to get the gRNA locus with two gRNAs for the target gene. This gRNA locus was then assembled with the backbone amplified from pGM‐xBCD to get the plasmid for the target gene. The plasmid was transformed into donor E. coli CA434 (or HB101/pRK24) and introduced into the recipient microbe via conjugation. The transconjugants harboring the CRISPRi‐dCpf1 plasmid are identified by antibiotic selection, and the gene knockdown is validated by qPCR of the target gene or other readout like LC‐MS measurement of metabolites.
Keynotes: To transfer plasmids containing the CRISPRi‐dCpf1 machinery into the target strains, in the step of plating conjugation cells onto agar plates with selective antibiotics, it is recommended to plate all 300 µL scraped‐cell suspension in PBS onto three plates, because the conjugation efficiency will decrease when the CRISPRi‐dCpf1 machinery is introduced into plasmids to make these plasmids bigger.
Potential issues and solutions: In some cases, the knockdown efficiency is low because the CRISPRi‐dCpf1 system has off‐target effects. To address this problem, one option is to test several different gRNA designs (construct different plasmids containing different gRNAs); the other option is to construct multiple gRNAs containing plasmids, an extended version of our current duplex gRNA design, introducing multiple gRNAs, like four gRNAs, would reduce the off‐target effects and enhance the suppressive efficiency of the CRISPRi‐dCpf1 system.
Experimental results interpretation: To determine the suppressive efficiency of target genes by the CRISPRi‐dCpf1 system, the expression of target genes is quantified by qPCR and normalized to reference 16s rRNA gene of the strain. In the case the introduced lacZα gene is the target gene, the strain harboring plasmid pGM‐xBCL (carrying CRISPRi‐dCpf1 + lacZα) serves as the control, and expression of the lacZα gene in the strain harboring plasmid pGM‐xBCF (carrying CRISPRi‐dCpf1 + lacZα + lacZα targeting gRNAs) is normalized to the expression of lacZα gene in control (as shown in Figure 3F). In the case, where a specific gene on the genome of the strain of interest is the target gene, the strain harboring plasmid pGM‐xBCD (carrying CRISPRi‐dCpf1) serves as the control, and expression of the target gene in the strain harboring plasmids that carry CRISPRi‐dCpf1 + gene targeting gRNAs is normalized to the expression of the target gene in control.
Developing a 16s‐tron strategy for Clostridia microbes
Group II intron‐based genetic tools were also developed for nonmodel gut Clostridia microbes. Group II intron was selected because it facilitates the insertion of retrotransposition‐activated markers (RAMs) into the targeted genome sites [15]. This targeted insertion does not induce lethal chromosomal double‐strand breaks (as initiated by Cas9) that need to be repaired by HR.
As in Bacteroidia microbes, conserved bacterial 16s rRNA genes of Clostridia strains were selected as a target gene to develop genetic manipulation tools without prior knowledge of their genome sequences. Multiple sequence alignment using 16s rRNAs of Clostridia that can uptake plasmids was performed to identify a highly conserved sequence that can be targeted by Group II intron. Then, the intron targeting and design tool on the ClosTron website (https://clostron.com/intron-design-tool) was used to design the Group II introns targeting the conserved 16s sequence. For each recipient Clostridia microbe, the designed 16s‐targeting Group II intron (16s‐tron) was assembled with their compatible rep oris and antibiotic RAM and transported into the recipient Clostridia microbes; the RAM provides antibiotic resistance only upon integration into the Clostridia chromosome.
Materials and devices
Primer star DNA polymerase (Takara, Cat# R045), Blue sapphire DNA polymerase (Takara, Cat# RR350), Plasmid Midiprep Kit (Zymo Research, Cat# D4201), DNA Clean and Concentrator (Zymo Research, Cat# D4003), Tryptic Soy Agar (BD, Cat# 236950), Brain Heart Infusion Agar (BD, Cat# 241830), CBA (BD, Cat# 279240), Horse blood (Hemostat Laboratories, Cat# 637291), LB broth (BD, Cat# BP1426), glycerol (Fisher Bioreagents, Cat# BP229), PBS (Gibco, Cat# 10010‐031), centrifuge, PCR amplifier, tetracycline (GoldBio, Cat# T‐101‐25), chloramphenicol (VWR, Cat# 0230), d‐cycloserine (D) (TCI, Cat# C1189), gentamicin (G) (GoldBio, Cat# G‐400‐25), kanamycin (K) (GoldBio, Cat# K‐120‐25), thiamphenicol (Thiam) (Acros Organics, Cat# 455450250), erythromycin (VWR, Cat# 0219), spectinomycin (Cat# BML‐A281‐0010), anaerobic chamber, aerobic incubator, electroporation system, Thermo Scientific Nanodrop 2000, Gibson Assembly Cloning Kit (NEB, Cat# E5510S), Quick DNA fungal/bacterial kit (Zymo Research, Cat# D6005), and ultralow temperature freezer.
Vector assembly
Two sets of plasmids (1) pGM‐xCAQ (pGM‐ACAQ, BCAQ, CCAQ, DCAQ, ECAQ, FCAQ, GCAQ, HCAQ, and ICAQ) whose conjugation‐selection marker is catP, and RAM is ermB, and (2) plasmids pGM‐xCBQ (pGM‐ACBQ, DCBQ, ECBQ, FCBQ, HCBQ, and ICBQ) whose conjugation‐selection marker is catP, and RAM is aad9, were generated to test the 16s‐tron strategy for Clostridia microbes [2] (Figure 4A–C). To target other specific genes in Clostridia strains, the introns for the target gene (~300 bp, Figure 4D) could be designed using the ClosTron website and synthesized and assembled with the backbone amplified from pGM‐xCAQ to get the plasmid for the target gene.
Figure 4.

Development of a 16s‐tron genetic manipulation strategy for Clostridia microbes. (A) Schematics of the starting Group II intron plasmid pGM‐BCAR‐001, which was previously assembled for the insertion of one target gene in the genome of Clostridium sporogenes ATCC 15579 (shown as “target intron”), with catP as the conjugation‐selection marker and ermB as the retrotransposition‐activated marker (RAM). G+ rep ori, Gram‐positive replication origin. (B, C) Schematics of the set of plasmids pGM‐ACAQ–pGM‐ICAQ and pGM‐ACBQ–pGM‐ICBQ carrying the 16s‐targeting Group II intron (16s‐tron), which mediate integration of the RAM into the targeted 16s rRNA genes, with catP as the conjugation‐selection marker, ermB (B) and aad9 (C) as the RAM. (D) Diagnostic PCR strategy to validate the 16s‐tron RAM integration into the 16s rRNA genes of targeted Clostridia microbes. The DiagF is the sequence on the RAM, which will not bind to the genome. The DiagR binds to the genome and will not bind to the Group II intron plasmid. There will be a PCR product of 2.0–2.5 kb as designed for colonies that have integrated the RAM, whereas no PCR product will be found for control colonies. PCR, polymerase chain reaction; rRNA, ribosomal RNA.
Keynotes: Different combinations of conjugation‐selection markers and RAM could be selected and used to replace the antibiotic markers in those established plasmids.
Introduction of the assembled 16s‐tron vectors and selection of the RAM‐integrated mutants
Using the strain Blautia luti DSM 14534 (S54) as an example, the plasmid pGM‐FCAQ was transformed into chemically competent E. coli CA434. Then E. coli CA434 harboring plasmid pGM‐FCAQ was conjugated to S54. The transconjugants were picked and restreaked onto a TSAB agar plate supplemented with d‐cycloserine (250 µg/mL) + thiamphenicol (15 µg/mL). Then, three single colonies were cultivated into 1 mL Mega liquid broth supplied with 15 µg/mL thiamphenicol and 250 µg/mL d‐cycloserine. After 24–36 h, 50 µL of cultures were spread onto TSAB agar plates supplemented with 250 µg/mL d‐cycloserine and 10 µg/mL erythromycin. The transconjugants typically appeared after 36–48 h. Eight colonies were picked to inoculate 3 mL Mega liquid broth supplemented with 250 µg/mL d‐cycloserine and 10 µg/mL erythromycin. After 24–36 h, genomic DNA was extracted using Quick DNA fungal/bacterial kit (Zymo Research) and diagnostic PCR was performed using primers 16s_tron_diagR_v4 + 16s_1391R + 16s_1391R_3to5 (with 16s_tron_diagR_v4 binding the integrated intron part and 16s_1391R + 16s_1391R_3to5 binding the target 16s site, only colonies that undergo RAM integration will have the band of ~2.5 kb) (Figure 4D).
This Group II intron strategy also readily applies to other genes of interest in Clostridia microbes. To mutate a target gene in Clostridia strains, the introns for the target gene (~300 bp, Figure 4D) are designed using the design tool on the ClosTron website and synthesized. The synthesized fragment was assembled with the backbone amplified from the set of plasmids pGM‐xCAQ with a compatible rep ori and antibiotic marker to get the plasmid for mutating the target gene. The plasmid was transferred into donor E. coli CA434 (or HB101/pRK24) and introduced into the recipient microbe via conjugation. The transconjugants harboring the plasmid are identified by antibiotic selection with d‐cycloserine and thiamphenicol. Then, colonies are cultivated into liquid broth with d‐cycloserine and thiamphenicol, and 50 µL of cultures are spread onto agar plates supplemented with d‐cycloserine and erythromycin (or other antibiotic markers to select for RAM insertion). The insertion mutants are validated by diagnostic PCR and sequencing as with 16s rRNA.
Keynotes: The Group II intron‐based plasmids are about 10 kb, which is much bigger than the plasmids used for the screening of plasmid uptake and will reduce the conjugation efficiency, in the step of plating conjugation cells onto agar plates with selective antibiotics, it is better to plate all 300 µL scraped‐cell suspension in PBS onto three different plates (100 µL onto each plate).
Potential issues and solutions: For some strains, the efficiency of the RAM selection step is low, and the above‐mentioned plating volume (50 µL) is not enough, in this case, more liquid could be plated onto the RAM selection plates, and is recommended to plate several plates.
Experimental results interpretation: For the result of diagnostic PCR of the transconjugants up on RAM selection, as shown in Figure 4D, because the forward diagnostic primer binds the sequence on the plasmid and the reverse diagnostic primer binds the sequence on the genome, only transconjugants that undergo the expected integration would have the 2.5 kb PCR product; WT strain or transconjugants that undergo the unexpected insertion would not have the 2.5 kb PCR product.
METHODS
Configuration methods and formulas for key solution reagents and medium is available in the Supporting Information.
SUMMARY
Many gut microbiota genes are associated with diseases like inflammatory bowel disease and colon cancer [1, 2, 3, 4, 5, 6], yet it is challenging to causally dissect their contributions at the molecular level because most gut commensals are nonmodel and genetically intractable. The genetic manipulation methods we introduce here provide an efficient and potentially generalizable microbiota genetic tool screening pipeline to screen nonmodel gut commensals and establish their tractable genetic systems on a large scale.
The bacterial 16s rRNA genes have long been used to reconstruct phylogenies and assess microbiome diversity. Since the Bacteroidia has relatively higher efficiency in HR, their highly conserved 16s rRNA gene could instead serve as an “archery target” to be inserted by the introduced suicide vector through a single crossover. Likewise, the 16s rRNA genes could also serve as a universal target in Clostridia microbes to be integrated by the 16s‐targeting Group II intron (16s‐tron) plasmid containing compatible rep oris and antibiotic RAM. Furthermore, CRISPR machinery targeting lacZα transcription in the introduced plasmid (CRISPRi‐lacZα system) could also be applied to establish the genetic tools in nonmodel Clostridia strains.
The pipeline we provide has three notable features. First, by targeting the 16s rRNA gene or assembling a CRISPRi‐dCpf1 lacZα system, the genetic systems could be built in gut bacteria without prior knowledge of their genome information. Second, without the “tune and test” process, the pipeline builds tractable genetic toolsets for multiple nonmodel Bacteroidia and Clostridia within weeks. Third, the pGM vectors are modular, and different genetic components, such as chimeric‐16s, rep oris, tagging markers like green fluorescent protein, or other nonnative genes of interesting biological function, can be switched/combined and introduced into these nonmodel gut commensals. All three features suggest the potential of the pipeline as a high‐throughput genetics screening and manipulating platform for the human gut microbiome.
Despite these advanced features, our strategies have limitations. First, the Bacteroidia genes are mutated via single crossover integration which sometimes leads to only partial dysfunction of their proteins, and this single crossover integration strategy would not work well when the size of the target gene is too small. In these cases, double crossover design could serve as a backup plan, with the left and right flanks of the target gene used for the single crossover, and double crossover comes later to get the expected knockout, our single crossover integration screening protocol can help pave the way for developing marker recycling system via double crossover in Bacteroides. Second, the CRISPRi‐dCpf1 system has off‐target effects; several gRNAs in the dCpf1 system might need to be tested to efficiently repress the target gene. Multiple gRNA design, an extended version of our current duplex gRNA design, may help overcome this limitation. Introducing multiple gRNAs, like four gRNAs, would reduce the off‐target effects and enhance the suppressive efficiency of the CRISPRi‐dCpf1 system [32, 33].
Although all the strategies described above are developed to screen and establish genetic manipulation systems in Bacteroidia and Clostridia strains on a relatively large scale, our protocol is applicable to microbes from other Phyla except Bacteroidia and Clostridia. For example, the single crossover integration strategy for Bacteroidia microbes using suicide plasmid works well in other Gram‐negative gut microbes. We have proved that suicide plasmid targeting strain‐specific 16s rRNA genes can be integrated into the targeted site of the genome of other Gram‐negative gut microbes, such as Fusobacterium gastrosuis DSM 101753, Fusobacterium nucleatum ATCC 25751, ATCC 10953, ATCC 23726, Klebsiella oxytoca DSM 29614, DSM 5175, DSM 7342, Proteus mirabilis ATCC 35659, and Proteus vulgaris DSM 3265. Also, the strategy of screening compatible rep ori‐harboring plasmids works in other Gram‐positive gut microbes. In Gram‐positive gut microbes like Bifidobacterium catenulatum DSM 16992 and several Enterococcus faecalis strains, we screened the compatible rep ori‐harboring plasmids and showed that the CRISPRi‐dCpf1 lacZα system worked well in those strains [34]. Furthermore, these strategies are readily applicable to other genes of interest (instead of 16s rRNA or lacZα, shown as “target genes” in Figures 1, 3, and 4) in other microbes, and the feasibility and reliability of the genetic manipulation strategies described here have been verified by multiple studies [34, 35]. This high‐throughput and generalizable protocol will greatly facilitate the molecular mechanism investigation of gut microbiota‐host interaction from the following aspects: (1) development of genetic tools in nonmodel bacteria that may be physiologically important, (2) precise manipulation of microbiota genes to assess their effect on host metabolism and biology, (3) disclosure of biosynthesis of microbiota‐derived metabolites like deoxycholic acid, and (4) stimulation of new strategies to engineer microbiota at the single gene level.
AUTHOR CONTRIBUTIONS
Chun‐Jun Guo and Wen‐Bing Jin conceived the protocol and designed the experiments. Wen‐Bing Jin performed the experiments and analyzed the data. Wen‐Bing Jin wrote the manuscript. Chun‐Jun Guo revised the manuscript and supervised this project. All authors have read the final manuscript and approved it for publication.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Supporting information
Table S1. Primers used in this protocol.
Supporting information.
ACKNOWLEDGMENTS
We thank Dr. Michael Fischbach, Dr. Justin Sonnenburg, and an anonymous healthy human fecal donor for their contribution to some bacterial strains we have used in this protocol. We also thank members of the Guo Group for helpful suggestions and comments on the manuscript. DNA sequencing was performed by Genewiz. The graphical abstract was created using BioRender.
Jin, Wen‐Bing , and Guo Chun‐Jun. 2024. “Genetic Manipulations of Nonmodel Gut Microbes.” iMeta 3, e216. 10.1002/imt2.216IF: 33.2 Q1
Contributor Information
Wen‐Bing Jin, Email: wej4002@med.cornell.edu.
Chun‐Jun Guo, Email: cj@guo-group.org.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study. Supplementary materials (figures, tables, graphical abstract, slides, videos, Chinese translated version, and update materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.
REFERENCES
-
1.
Yachida, Shinichi
, Mizutani Sayaka, Shiroma Hirotsugu, Shiba Satoshi, Nakajima Takeshi, Sakamoto Taku, Watanabe Hikaru, et al. 2019. “Metagenomic and Metabolomic Analyses Reveal Distinct Stage‐Specific Phenotypes of the Gut Microbiota in Colorectal Cancer.” Nature Medicine
25: 968–976. 10.1038/s41591-019-0458-7IF: 50.0 Q1
[DOI] [PubMed] [Google Scholar]
1. 矢千田、真一、水谷沙耶香、城间弘次、柴智、中岛武、坂本拓、渡边光等。 2019。“宏基因组和代谢组学分析揭示了结直肠癌肠道微生物群的不同阶段特异性表型。”自然医学 25:968–976。 10.1038/s41591-019-0458-7 如果:50.0 Q1 [ DOI ] [ PubMed ] [ Google Scholar ] -
2.
Lloyd‐Price, Jason
, Arze Cesar, Ananthakrishnan Ashwin N., Schirmer Melanie, Avila‐Pacheco Julian, Poon Tiffany W., Andrews Elizabeth, et al. 2019. “Multi‐Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases.” Nature
569: 655–662. 10.1038/s41586-019-1237-9IF: 48.5 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
2. Lloyd‐Price, Jason、Arze Cesar、Ananthakrishnan Ashwin N.、Schirmer Melanie、Avila‐Pacheco Julian、Poon Tiffany W. 和 Andrews Elizabeth 等人,2019 年。“炎症性肠病中肠道微生物生态系统的多组学研究。”《自然》569: 655–662。10.1038/s41586-019-1237-9 如果:48.5 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
3.
Thomas, Andrew Maltez
, Manghi Paolo, Asnicar Francesco, Pasolli Edoardo, Armanini Federica, Zolfo Moreno, Beghini Francesco, et al. 2019. “Metagenomic Analysis of Colorectal Cancer Datasets Identifies Cross‐Cohort Microbial Diagnostic Signatures and a Link with Choline Degradation.” Nature Medicine
25: 667–678. 10.1038/s41591-019-0405-7IF: 50.0 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
3. Thomas、Andrew Maltez、Manghi Paolo、Asnicar Francesco、Pasolli Edoardo、Armanini Federica、Zolfo Moreno、Beghini Francesco 等。 2019。“结直肠癌数据集的宏基因组分析确定了跨队列微生物诊断特征以及与胆碱降解的联系。”自然医学 25:667–678。 10.1038/s41591-019-0405-7 如果:50.0 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
4.
Wirbel, Jakob
, Pyl Paul Theodor, Kartal Ece, Zych Konrad, Kashani Alireza, Milanese Alessio, Fleck Jonas S., et al. 2019. “Meta‐Analysis of Fecal Metagenomes Reveals Global Microbial Signatures That Are Specific for Colorectal Cancer.” Nature Medicine
25: 679–689. 10.1038/s41591-019-0406-6IF: 50.0 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
4. Wirbel, Jakob, Pyl Paul Theodor, Kartal Ece, Zych Konrad, Kashani Alireza, Milanese Alessio, Fleck Jonas S. 等,2019. “粪便宏基因组的荟萃分析揭示了结直肠癌特异性的全球微生物特征。”《自然医学》25: 679–689. 10.1038/s41591-019-0406-6 如果:50.0 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
5.
Zhou, Wenyu
, Sailani M. Reza, Contrepois Kévin, Zhou Yanjiao, Ahadi Sara, Leopold Shana R., Zhang Martin J., et al. 2019. “Longitudinal Multi‐Omics of Host–Microbe Dynamics in Prediabetes.” Nature
569: 663–671. 10.1038/s41586-019-1236-xIF: 48.5 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
5. 周文宇、Sailani M. Reza、Contrepois Kévin、周燕娇、Ahadi Sara、Leopold Shana R. 和 Zhang Martin J. 等人,2019 年。“糖尿病前期宿主-微生物动态的纵向多组学研究。”《自然》569: 663–671。10.1038/s41586-019-1236-x 如果:48.5 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
6.
Qin, Junjie
, Li Yingrui, Cai Zhiming, Li Shenghui, Zhu Jianfeng, Zhang Fan, Liang Suisha, et al. 2012. “A Metagenome‐Wide Association Study of Gut Microbiota in Type 2 Diabetes.” Nature
490: 55–60. 10.1038/nature11450IF: 48.5 Q1
[DOI] [PubMed] [Google Scholar]
6. 秦俊杰,李英锐,蔡志明,李生辉,朱剑锋,张帆,梁穗莎,等。 2012.“2 型糖尿病肠道微生物群的宏基因组范围关联研究”。自然 490:55–60。 10.1038/自然 11450 如果:48.5 Q1 [ DOI ] [ PubMed ] [ Google Scholar ] -
7.
Cho, Ilseung
, and Blaser Martin J.. 2012. “The Human Microbiome: At the Interface of Health and Disease.” Nature Reviews Genetics
13(4): 260–270. 10.1038/nrg3182IF: 52.0 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
7. Cho, Ilseung 和 Blaser Martin J.。2012 年。“人类微生物组:健康与疾病的交汇点。”《自然评论:遗传学》13(4): 260–270。10.1038/nrg3182 如果:52.0 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
8.
Lloyd‐Price, Jason
, Abu‐Ali Galeb, and Huttenhower Curtis. 2016. “The Healthy Human Microbiome.” Genome Medicine
8(1): 51. 10.1186/s13073-016-0307-yIF: 11.2 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
8. Lloyd‐Price, Jason, Abu‐Ali Galeb 和 Huttenhower Curtis。2016.“健康的人类微生物组。”基因组医学 8(1): 51。10.1186/s13073-016-0307-y 如果:11.2 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
9.
Taketani, Mao
, Zhang Jianbo, Zhang Shuyi, Triassi Alexander J., Huang Yu‐Ja, Griffith Linda G., and Voigt Christopher A.. 2020. “Genetic Circuit Design Automation for the Gut Resident Species Bacteroides thetaiotaomicron
.” Nature Biotechnology
38(8): 962–969. 10.1038/s41587-020-0468-5IF: 41.7 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
9. Taketani, Mao, Zhang Jianbo, Zhang Shuyi, Triassi Alexander J., Huang Yu‐Ja, Griffith Linda G., 和 Voigt Christopher A.. 2020. “肠道常驻菌种多形拟杆菌的遗传电路设计自动化。”《自然生物技术》38(8): 962–969. 10.1038/s41587-020-0468-5 如果:41.7 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
10.
Cañadas, Inés C.
, Groothuis Daphne, Zygouropoulou Maria, Rodrigues Raquel, and Minton Nigel P.. 2019. “RiboCas: A Universal CRISPR‐based Editing Tool for Clostridium
.” ACS Synthetic Biology
8: 1379–1390. 10.1021/acssynbio.9b00075IF: 3.9 Q1
[DOI] [PubMed] [Google Scholar]
10. Canadas、Inés C.、Groothuis Daphne、Zygouropoulou Maria、Rodrigues Raquel 和 Minton Nigel P.. 2019。“RiboCas:基于 CRISPR 的通用梭状芽胞杆菌编辑工具”。美国化学学会合成生物学 8:1379–1390。 10.1021/acssynbio.9b00075 如果:3.9 Q1 [ DOI ] [ PubMed ] [ Google Scholar ] -
11.
Guo, Chun‐Jun
, Allen Breanna M., Hiam Kamir J., Dodd Dylan, Van Treuren Will, Higginbottom Steven, Nagashima Kazuki, et al. 2019. “Depletion of Microbiome‐Derived Molecules in the Host Using Clostridium Genetics.” Science
366: eaav1282. 10.1126/science.aav1282IF: 45.8 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
11.Guo 、Chun‐Jun、Allen Breanna M.、Hiam Kamir J.、Dodd Dylan、Van Treuren Will、Higginbottom Steven、Nagashima Kazuki 等。 2019.“利用梭状芽胞杆菌遗传学消除宿主中微生物组衍生的分子。”科学 366:eaav1282。 10.1126/science.aav1282 如果:45.8 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
12.
Wu, Meng
, McNulty Nathan P., Rodionov Dmitry A., Khoroshkin Matvei S., Griffin Nicholas W., Cheng Jiye, Latreille Phil, et al. 2015. “Genetic Determinants of In Vivo Fitness and Diet Responsiveness in Multiple Human Gut Bacteroides
.” Science
350(6256): aac5992. 10.1126/science.aac5992IF: 45.8 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
12. Wu, Meng 、McNulty Nathan P.、Rodionov Dmitry A.、Khoroshkin Matvei S.、Griffin Nicholas W.、Cheng Jiye、Latreille Phil 等人,2015 年。“多种人类肠道拟杆菌体内适应性和饮食反应性的遗传决定因素。”Science 350(6256): aac5992。10.1126/science.aac5992 如果:45.8 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
13.
Mimee, Mark
, Tucker Alex C., Voigt Christopher A., and Lu Timothy K.. 2015. “Programming a Human Commensal Bacterium, Bacteroides thetaiotaomicron, to Sense and Respond to Stimuli In the Murine Gut Microbiota.” Cell Systems
1: 62–71. 10.1016/j.cels.2015.06.001IF: 7.7 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
13. Mimee, Mark、Tucker Alex C.、Voigt Christopher A. 和 Lu Timothy K.,2015. “对人类共生菌 Bacteroides thetaiotaomicron 进行编程,使其感知并响应小鼠肠道菌群中的刺激。” Cell Systems 1: 62–71. 10.1016/j.cels.2015.06.001 如果:7.7 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
14.
Goodman, Andrew L.
, McNulty Nathan P., Zhao Yue, Leip Douglas, Mitra Robi D., Lozupone Catherine A., Knight Rob, and Gordon Jeffrey I.. 2009. “Identifying Genetic Determinants Needed to Establish a Human Gut Symbiont in Its Habitat.” Cell Host & Microbe
6: 279–289. 10.1016/j.chom.2009.08.003IF: 18.7 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
14. Goodman, Andrew L.、McNulty Nathan P.、Zhao Yue、Leip Douglas、Mitra Robi D.、Lozupone Catherine A.、Knight Rob 和 Gordon Jeffrey I.,2009 年。“确定在人体肠道共生体栖息地建立所需的遗传决定因素。”《细胞宿主与微生物》6: 279–289。10.1016/j.chom.2009.08.003 如果:18.7 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
15.
Heap, John T.
, Pennington Oliver J., Cartman Stephen T., Carter Glen P., and Minton Nigel P.. 2007. “The ClosTron: A Universal Gene Knock‐Out System for the Genus Clostridium
.” Journal of Microbiological Methods
70: 452–464. 10.1016/j.mimet.2007.05.021IF: 1.9 Q3
[DOI] [PubMed] [Google Scholar]
15. Heap, John T.、Pennington Oliver J.、Cartman Stephen T.、Carter Glen P. 和 Minton Nigel P.,2007. “ClosTron: 梭菌属的通用基因敲除系统。”《微生物方法杂志》70: 452–464。10.1016/j.mimet.2007.05.021 如果:1.9 Q3 [ DOI ] [ PubMed ] [ Google Scholar ] -
16.
Salyers, Abigail A.
, Shoemaker Nadja, Cooper Andrew, D'Elia John, and Shipman Joseph A.. 1999. “8 Genetic Methods for Bacteroides Species.” Methods in Microbiology
29: 229–249. 10.1016/S0580-9517(08)70119-3
[DOI] [Google Scholar]
16. Salyers, Abigail A.、Shoemaker Nadja、Cooper Andrew、D'Elia John 和 Shipman Joseph A.。1999 年。“8 种拟杆菌属的遗传方法。”《微生物学方法》29: 229–249。10.1016/S0580-9517(08)70119-3 [ DOI ] [ Google Scholar ] -
17.
Whitaker, Weston R.
, Shepherd Elizabeth Stanley, and Sonnenburg Justin L.. 2017. “Tunable Expression Tools Enable Single‐Cell Strain Distinction in the Gut Microbiome.” Cell
169: 538–546.e12. 10.1016/j.cell.2017.03.041IF: 42.5 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
17. Whitaker, Weston R.、Shepherd Elizabeth Stanley 和 Sonnenburg Justin L.,2017 年。“可调表达工具可区分肠道微生物组中的单细胞菌株。”Cell 169: 538–546.e12。10.1016/j.cell.2017.03.041 如果:42.5 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
18.
Lim, Bentley
, Zimmermann Michael, Barry Natasha A., and Goodman Andrew L.. 2017. “Engineered Regulatory Systems Modulate Gene Expression of Human Commensals in the Gut.” Cell
169: 547–558.e15. 10.1016/j.cell.2017.03.045IF: 42.5 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
18. Lim, Bentley、Zimmermann Michael、Barry Natasha A. 和 Goodman Andrew L.,2017 年。“工程调控系统调节人类肠道共生菌的基因表达。”Cell 169: 547–558.e15。10.1016/j.cell.2017.03.045 如果:42.5 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
19.
García‐Bayona, Leonor
, and Comstock Laurie E.. 2019. “Streamlined Genetic Manipulation of Diverse Bacteroides and Parabacteroides Isolates from the Human Gut Microbiota.” mBio
10(4): e01762–19. 10.1128/mbio.01762-19IF: 4.7 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
19. García-Bayona、Leonor 和 Comstock Laurie E.. 2019。“从人类肠道微生物群中分离出多种拟杆菌和副拟杆菌的简化遗传操作。” mBio 10(4):e01762–19。 10.1128/mbio.01762-19 如果:4.7 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
20.
Bencivenga‐Barry, Natasha A.
, Lim Bentley, Herrera Carmen M., Trent M. Stephen, and Goodman Andrew L.. 2020. “Genetic Manipulation of Wild Human Gut Bacteroides
.” Journal of Bacteriology
202(3): e00544–19. 10.1128/jb.00544-19IF: 3.0 Q3
[DOI] [PMC free article] [PubMed] [Google Scholar]
20. Bencivenga‐Barry, Natasha A., Lim Bentley, Herrera Carmen M., Trent M. Stephen 和 Goodman Andrew L. . 2020. “野生人类肠道拟杆菌的遗传操作。”《细菌学杂志》202(3): e00544–19. 10.1128/jb.00544-19 如果:3.0 Q3 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
21.
Martens, Eric C.
, Chiang Herbert C., and Gordon Jeffrey I.. 2008. “Mucosal Glycan Foraging Enhances Fitness and Transmission of a Saccharolytic Human Gut Bacterial Symbiont.” Cell Host & Microbe
4: 447–457. 10.1016/j.chom.2008.09.007IF: 18.7 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
21. Martens, Eric C.、Chiang Herbert C. 和 Gordon Jeffrey I.,2008. “粘膜聚糖觅食增强糖分解人类肠道细菌共生体的适应性和传播。”《细胞宿主与微生物》4: 447–457。10.1016/j.chom.2008.09.007 如果:18.7 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
22.
Garnier, Thierry
, and Cole Stewart T.. 1988. “Identification and Molecular Genetic Analysis of Replication Functions of the Bacteriocinogenic Plasmid pIP404 From Clostridium perfringens
.” Plasmid
19: 151–160. 10.1016/0147-619X(88)90053-4IF: 2.2 Q3
[DOI] [PubMed] [Google Scholar]
22. Garnier, Thierry 和 Cole Stewart T.,1988 年。“ 产气荚膜梭菌产细菌素质粒 pIP404 的鉴定及其复制功能的分子遗传学分析。”质粒 19: 151–160。10.1016/0147-619X(88)90053-4 如果:2.2 Q3 [ DOI ] [ PubMed ] [ Google Scholar ] -
23.
Trieu‐Cuot, Patrick
, Carlier Cécile, Poyart‐Salmeron Claire, and Courvalin Patrice. 1991. “Shuttle Vectors Containing a Multiple Cloning Site and a La &Amp; a Escherichia coli to Gram‐Positive Bacteria Gene for Conjugal Transfer of DNA from Escherichia coli to Gram‐Positive Bacteria.” Gene
102: 99–104. 10.1016/0378-1119(91)90546-NIF: 2.4 Q3
[DOI] [PubMed] [Google Scholar]
23. Trieu-Cuot, Patrick, Carlier Cécile, Poyart-Salmeron Claire 和 Courvalin Patrice。1991. “含有多克隆位点和 La 和大肠杆菌至革兰氏阳性菌基因的穿梭载体,用于将 DNA 从大肠杆菌接合转移到革兰氏阳性菌。” 基因 102: 99–104。10.1016/0378-1119(91)90546-N 如果:2.4 Q3 [ DOI ] [ PubMed ] [ Google Scholar ] -
24.
Tardif, C.
, Maamar H., Balfin M., and Belaich J. P.. 2001. “Electrotransformation Studies in Clostridium cellulolyticum
.” Journal of Industrial Microbiology and Biotechnology
27: 271–274. 10.1038/sj.jim.7000081IF: 3.2 Q2
[DOI] [PubMed] [Google Scholar]
24. Tardif, C.、Maamar H.、Balfin M. 和 Belaich JP。2001. “ Clostridium cellulolyticum 的电转化研究。”《工业微生物与生物技术杂志》27: 271–274。10.1038/sj.jim.7000081 如果:3.2 Q2 [ DOI ] [ PubMed ] [ Google Scholar ] -
25.
McAllister, Kathleen N.
, Bouillaut Laurent, Kahn Jennifer N., Self William T., and Sorg Joseph A.. 2017. “Using CRISPR‐Cas9‐mediated Genome Editing to Generate C. difficile Mutants Defective in Selenoproteins Synthesis.” Scientific Reports
7: 14672. 10.1038/s41598-017-15236-5IF: 3.9 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
25. McAllister, Kathleen N.、Bouillaut Laurent、Kahn Jennifer N.、Self William T. 和 Sorg Joseph A.,2017 年。“利用 CRISPR-Cas9 介导的基因组编辑生成硒蛋白合成缺陷的艰难梭菌突变体。”《科学报告》7: 14672。10.1038/s41598-017-15236-5 如果:3.9 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
26.
Zetsche, Bernd
, Gootenberg Jonathan S., Abudayyeh Omar O., Slaymaker Ian M., Makarova Kira S., Essletzbichler Patrick, Volz Sara E., et al. 2015. “Cpf1 Is a Single RNA‐guided Endonuclease of a Class 2 CRISPR‐Cas System.” Cell
163: 759–771. 10.1016/j.cell.2015.09.038IF: 42.5 Q1
[DOI] [PMC free article] [PubMed] [Google Scholar]
26. Zetsche, Bernd、Gootenberg Jonathan S.、Abudayyeh Omar O.、Slaymaker Ian M.、Makarova Kira S.、Essletzbichler Patrick 和 Volz Sara E. 等人,2015. “Cpf1 是一种 2 类 CRISPR-Cas 系统的单 RNA 引导内切酶。” Cell 163: 759–771。10.1016/j.cell.2015.09.038 如果:42.5 Q1 [ DOI ] [ PMC free article ] [ PubMed ] [ Google Scholar ] -
27.
Kim, Seong Keun
, Kim Haseong, Ahn Woo‐Chan, Park Kwang‐Hyun, Woo Eui‐Jeon, Lee Dae‐Hee, and Lee Seung‐Goo. 2017. “Efficient Transcriptional Gene Repression by Type V‐A CRISPR‐Cpf1 from Eubacterium eligens
.” ACS Synthetic Biology
6: 1273–1282. 10.1021/acssynbio.6b00368IF: 3.9 Q1
[DOI] [PubMed] [Google Scholar]
27. Kim, Seong Keun、Kim Haseong、Ahn Woo-Chan、Park Kwang-Hyun、Woo Eui-Jeon、Lee Dae-Hee 和 Lee Seung-Goo。2017. “ 真杆菌中 V-A 型 CRISPR-Cpf1 基因高效转录抑制。”ACS 合成生物学 6: 1273–1282。10.1021/acssynbio.6b00368 如果:3.9 Q1 [ DOI ] [ PubMed ] [ Google Scholar ] - 28. Tang, Xu , Lowder Levi G., Zhang Tao, Malzahn Aimee A., Zheng Xuelian, Voytas Daniel F., Zhong Zhaohui, et al. 2017. “Correction: A CRISPR–Cpf1 System for Efficient Genome Editing and Transcriptional Repression in Plants.” Nature Plants 3(3): 17103. 10.1038/nplants.2017.103IF: 13.6 Q1 [DOI] [PubMed] [Google Scholar]
- 29. Hong, Wei , Zhang Jie, Cui Guzhen, Wang Luxin, and Wang Yi. 2018. “Multiplexed CRISPR‐Cpf1‐mediated Genome Editing in Clostridium difficile Toward the Understanding of Pathogenesis of C. difficile Infection.” ACS Synthetic Biology 7: 1588–1600. 10.1021/acssynbio.8b00087IF: 3.9 Q1 [DOI] [PubMed] [Google Scholar]
- 30. Hur, Junho K. , Kim Kyoungmi, Been Kyung Wook, Baek Gayoung, Ye Sunghyeok, Hur Junseok W., Ryu Seuk‐Min, Lee Youn Su, and Kim Jin‐Soo. 2016. “Targeted Mutagenesis in Mice by Electroporation of Cpf1 Ribonucleoproteins.” Nature Biotechnology 34(8): 807–808. 10.1038/nbt.3596IF: 41.7 Q1 [DOI] [PubMed] [Google Scholar]
- 31. Zhang, Xiaochun , Wang Jingman, Cheng Qiuxiang, Zheng Xuan, Zhao Guoping, and Wang Jin. 2017. “Multiplex Gene Regulation by CRISPR‐ddCpf1.” Cell Discov 3: 17018. 10.1038/celldisc.2017.18IF: 12.5 Q1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Breunig, Christopher T. , Durovic Tamara, Neuner Andrea M., Baumann Valentin, Wiesbeck Maximilian F., Köferle Anna, Götz Magdalena, Ninkovic Jovica, and Stricker Stefan H.. 2018. “One Step Generation of Customizable gRNA Vectors for Multiplex CRISPR Approaches Through String Assembly gRNA Cloning (STAgR).” PLoS ONE 13: e0196015. 10.1371/journal.pone.0196015IF: 2.6 Q2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim, Won‐Nyeong , Kim Hye‐Jeong, Chung Young‐Soo, and Kim Hyun‐Uk. 2021. “Construction of Multiple Guide RNAs in CRISPR/Cas9 Vector Using Stepwise or Simultaneous Golden Gate Cloning: Case Study for Targeting the FAD2 and FATB Multigene in Soybean.” Plants 10: 2542. 10.3390/plants10112542IF: 4.1 Q1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jin, Wen‐Bing , Li Ting‐Ting, Huo Da, Qu Sophia, Li Xin V., Arifuzzaman Mohammad, Lima Svetlana F., et al. 2022. “Genetic Manipulation of Gut Microbes Enables Single‐Gene Interrogation in a Complex Microbiome.” Cell 185: 547–562.e22. 10.1016/j.cell.2021.12.035IF: 42.5 Q1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang, Pengjie , Tian Jinzhong, Zhang Lu, Zhang Hui, Yang Gaohua, Ren Yimeng, Fang Jingyuan, Gu Yang, and Jiang Weihong. 2024. “A Toolbox for Genetic Manipulation in Intestinal Clostridium Symbiosum.” Synthetic and Systems Biotechnology 9: 43–54. 10.1016/j.synbio.2023.12.005IF: 4.4 Q1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used in this protocol.
Supporting information.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study. Supplementary materials (figures, tables, graphical abstract, slides, videos, Chinese translated version, and update materials) may be found in the online DOI or iMeta Science http://www.imeta.science/.