
Abstract 摘要
Normal circulatory function is a key determinant of disease-free life expectancy (healthspan). Indeed, pathologies affecting the cardiovascular system, which are growing in prevalence, are the leading cause of global morbidity, disability and mortality, whereas the maintenance of cardiovascular health is necessary to promote both organismal healthspan and lifespan. Therefore, cardiovascular ageing might precede or even underlie body-wide, age-related health deterioration. In this Review, we posit that eight molecular hallmarks are common denominators in cardiovascular ageing, namely disabled macroautophagy, loss of proteostasis, genomic instability (in particular, clonal haematopoiesis of indeterminate potential), epigenetic alterations, mitochondrial dysfunction, cell senescence, dysregulated neurohormonal signalling and inflammation. We also propose a hierarchical order that distinguishes primary (upstream) from antagonistic and integrative (downstream) hallmarks of cardiovascular ageing. Finally, we discuss how targeting each of the eight hallmarks might be therapeutically exploited to attenuate residual cardiovascular risk in older individuals.
正常的循环功能是无病生存寿命(健康寿命)的关键决定因素。实际上,影响心血管系统的疾病日益普遍,是全球发病、残疾和死亡的主要原因,而维持心血管健康对于延长机体健康寿命和寿命都是必要的。因此,心血管衰老可能先于甚至导致全身性的、与年龄相关的健康恶化。 在本综述中,我们认为八个分子特征是心血管衰老的共同决定因素,即巨自噬功能障碍、蛋白稳态丧失、基因组不稳定性(特别是具有不确定潜能的克隆性造血)、表观遗传改变、线粒体功能障碍、细胞衰老、神经激素信号失调和炎症。 我们还提出了一个层级顺序,区分了心血管老化的主要(上游)标志与拮抗性和整合性(下游)标志。最后,我们讨论了如何以针对这八个标志的治疗手段来降低老年人的残余心血管风险。
Key points 要点
-
Age is one of the strongest determinants of cardiovascular health.
年龄是决定心血管健康的最重要因素之一。 -
The cardiovascular system operates under constant mechanical and metabolic stress, which progressively increases the risk of dysfunction at the molecular, cellular and whole-organ levels with increasing age.
心血管系统在持续的机械和代谢压力下运作,随着年龄的增长,分子、细胞和整个器官水平的功能障碍风险逐渐增加。 -
Disabled macroautophagy, loss of proteostasis, genomic instability, epigenetic alterations, mitochondrial dysfunction, cell senescence, dysregulated neurohormonal signalling and inflammation emerge as common molecular hallmarks of cardiovascular ageing.
巨自噬功能障碍、蛋白稳态丧失、基因组不稳定、表观遗传改变、线粒体功能障碍、细胞衰老、神经激素信号失调和炎症是心血管衰老的常见分子标志。 -
The hallmarks of cardiovascular ageing are strongly intertwined, and accentuation or attenuation of individual hallmarks affects most, if not all, the others.
心血管衰老的标志是紧密交织的,单个标志的加重或减轻会影响大多数(如果不是全部)其他标志。 -
Targeting the hallmarks of cardiovascular ageing holds promise for the treatment of major cardiovascular disorders and to reduce residual cardiovascular risk beyond the management of conventional risk factors.
靶向心血管衰老的特征有望用于治疗主要心血管疾病,并在传统危险因素管理之外降低残余心血管风险。
Similar content being viewed by others
其他人正在查看的类似内容

Introduction 导言
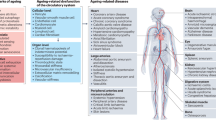
The human cardiovascular system is composed of multiple distinct cell types, including endothelial cells, smooth muscle cells, cardiomyocytes, fibroblasts and immune cells, which work harmoniously together to maintain sufficient blood supply to, and waste disposal from, every cell in the body. Notably, most cardiovascular cell types are highly specialized and need to function under hostile conditions, characterized by unremitting mechanical pressure and shear stress. However, the acquisition of unique functions (as exemplified by cardiomyocytes, the sole contractile cell type in the heart) comes at the expense of cell cycle withdrawal. As such, these terminally differentiated cells, which are densely packed with mitochondria and sarcomeres, have limited proliferative capacity and must endure the stressful working conditions of the cardiovascular system for the whole lifespan of the individual1,2,3. Although ageing inexorably erodes the physiological functions of every cell type in the body, cardiomyocytes and other cardiovascular cells might be at particularly elevated risk of age-related dysfunction and failure. Indeed, with ageing, the heart, vessels and microcirculation all undergo substantial structural and functional remodelling4,5,6,7,8 (Box 1) as the cellular composition of the cardiovascular system is altered (Fig. 1). Therefore, it is not surprising that age is among the strongest predictors of cardiovascular health9. Moreover, despite outstanding progress in the medical management of cardiovascular risk factors, such as hypertension and dyslipidaemia10,11, pathologies affecting the cardiovascular system remain the leading cause of global morbidity, disability and mortality12.
人体心血管系统由多种不同的细胞类型组成,包括内皮细胞、平滑肌细胞、心肌细胞、成纤维细胞和免疫细胞,这些细胞和谐地协同工作,以维持足够的血液供应,并将体内每个细胞的废物排出。值得注意的是,大多数心血管细胞类型都是高度特化的,需要在恶劣的条件下发挥作用,其特点是持续不断的机械压力和剪切应力。 然而,获得独特功能(以心肌细胞为例,它是心脏中唯一具有收缩功能的细胞类型)的代价是细胞周期停止。因此,这些终末分化的细胞,其内部密布着线粒体和肌节,增殖能力有限,必须在个体的一生中承受心血管系统充满压力的工作条件 1,2,3 。 虽然衰老不可避免地会侵蚀体内每种细胞类型的生理功能,但心肌细胞和其他心血管细胞可能面临与年龄相关的机能障碍和衰竭的特别高的风险。事实上,随着年龄的增长,心脏、血管和微循环都会发生大量的结构和功能重塑 4,5,6,7,8 (框 1),因为心血管系统的细胞组成会发生改变(图 1)。 因此,年龄是心血管健康的最强预测指标之一,这一点不足为奇 9 。此外,尽管在心血管危险因素(如高血压和血脂异常)的医疗管理方面取得了显著进展 10,11 ,但影响心血管系统的疾病仍然是全球发病、残疾和死亡的主要原因 12 。
图 1:衰老对心血管系统细胞组成的影响。

a, The proportions of various cell types in the heart and aorta of mice aged 1–3 months, 18–21 months and 24–30 months. Reported values were derived using the microfluidic droplet method for single-cell analysis published in the Tabula Muris Senis (mouse ageing cell atlas)442. Values represent the average of 2–5 mice per group. b, Changes in the cellular composition of the human heart with increasing age. Values represent the mean of 7–10 donor hearts per group subjected to single-nucleus RNA sequencing analysis at the age of 11–39 years, 44–60 years or 61–75 years364. Of note, given that the values show only age-dependent changes for each species, direct comparisons between mice and humans cannot be drawn due to differences in the age groups, tissues (heart and aorta in mice versus heart only in humans) and applied techniques (single-cell and single-nucleus RNA sequencing in mice and humans, respectively). The exact proportion of a given cell type within the same species might also vary if measured with different techniques such as fluorescence-activated cell sorting442,443. A discussion of the findings and limitations of these techniques is given in Box 2.
a,1-3 个月、18-21 个月和 24-30 个月大的小鼠心脏和主动脉中各种细胞类型的比例。报告的值是使用 Tabula Muris Senis(小鼠衰老细胞图谱) 442 中发表的单细胞分析微流体液滴法得出的。数值代表每组 2-5 只小鼠的平均值。b,人类心脏的细胞组成随年龄增长而发生变化。 数值代表每组 7–10 个供体心脏的平均值,这些心脏在 11–39 岁、44–60 岁或 61–75 岁时接受了单细胞核 RNA 测序分析 364 。 值得注意的是,鉴于这些数值仅显示了每个物种的年龄依赖性变化,因此由于年龄组、组织(小鼠为心脏和主动脉,而人类仅为心脏)和应用技术(小鼠和人类分别为单细胞和单核 RNA 测序)的差异,无法直接比较小鼠和人类。 如果使用不同的技术(如荧光激活细胞分选 442,443 )进行测量,即使在同一物种内,特定细胞类型的确切比例也可能有所不同。关于这些技术的研究结果和局限性,请见方框 2 中的讨论。
In this Review, we speculate that the persistently high rates of cardiovascular disease and death, commonly referred to as ‘residual cardiovascular risk’13,14,15, are attributable to the absence of efficient interventions targeting the ageing process itself16. Indeed, even the sexual dimorphism in cardiovascular risk is increasingly being attributed to differences in the rates of ageing between men and women4,17,18. Intriguingly, experimental interventions designed specifically to delay cardiovascular ageing are sufficient to attenuate ageing in model organisms. For instance, genetic manipulation of cardiomyocyte insulin-like growth factor 1 (IGF1) signalling has been shown to extend both the healthspan and lifespan of mice19. Similarly, targeting vascular endothelial growth factor (VEGF) signalling prevents age-related microvascular attrition and effectively delays age-related pathologies across multiple organ systems, resulting in a prolonged lifespan of mice20. These examples underpin the central role of cardiovascular deterioration in systemic ageing and suggest that targeting the circulatory system is a viable entry point to promote healthy ageing at the organismal level.
在本综述中,我们推测心血管疾病和死亡率持续居高不下(通常被称为“残余心血管风险” 13,14,15 )的原因是缺乏针对衰老过程本身的有效干预措施 16 。事实上,心血管风险中的性别二态性也越来越归因于男性和女性衰老速度的差异 4,17,18 。 有趣的是,专门设计用于延缓心血管老化的实验干预足以减缓模型生物的老化。例如,心肌细胞胰岛素样生长因子 1(IGF1)信号通路的基因干预已被证明可以延长小鼠的健康寿命和寿命 19 。 同样地,靶向血管内皮生长因子(VEGF)信号传导可以预防与年龄相关的微血管衰退,并有效延缓多个器官系统中与年龄相关的病理,从而延长小鼠的寿命 20 。这些例子强调了心血管退化在全身衰老中的核心作用,并表明靶向循环系统是促进生物体层面健康衰老的可行切入点。
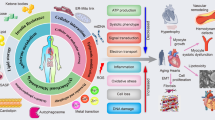
The premise of anti-ageing interventions relies on targeting the molecular mechanisms of ageing. However, many of the classical ‘hallmarks of ageing’ were originally discovered in invertebrate model organisms21. Furthermore, although these hallmarks were revised in 2023 with a focus on mammalian ageing22, their specific involvement in the dysfunction of major cardiovascular cell types during ageing remains to be appraised. In this Review, we propose eight molecular hallmarks as common denominators of ageing in the cardiovascular system (Fig. 2), based on three stringent criteria:
抗衰老干预的前提是靶向衰老的分子机制。然而,许多经典的“衰老标志”最初是在无脊椎动物模型生物中发现的 21 。此外,尽管这些标志在 2023 年进行了修订,重点关注哺乳动物的衰老 22 ,但它们在衰老过程中对主要心血管细胞类型功能障碍的具体影响仍有待评估。 在这篇综述中,我们基于三个严格的标准,提出了八个分子特征作为心血管系统衰老的共同决定因素(图 2):
-
The phenomenon must manifest in ageing hearts, vessels or both.
这种现象必须体现在衰老的心脏、血管或两者中。 -
Genetic manipulations promoting the hallmark must accelerate the signs of cardiovascular ageing.
促进这一特征的基因操作必须加速心血管衰老的迹象。 -
Experimental or therapeutic suppression of the hallmark should delay, halt or reverse cardiovascular ageing.
对这一特征的实验性或治疗性抑制应该延迟、阻止或逆转心血管衰老。
图 2:心血管衰老的标志。

The eight proposed hallmarks of ageing in the cardiovascular system are shown with their putative hierarchy. The ‘primary’ hallmarks progressively advance with age and underlie the deterioration of the genome, epigenome, proteome and organelles. The ‘antagonistic’ hallmarks reflect (mal)adaptive responses to damage, and the integrative hallmark ultimately precipitates the manifestation of cardiovascular disease.
心血管系统中八个已提出的衰老标志及其假定的层级结构如图所示。“主要”标志随年龄增长逐渐发展,并导致基因组、表观基因组、蛋白质组和细胞器的恶化。“拮抗性”标志反映了对损伤的(不良)适应性反应,而整合性标志最终促成了心血管疾病的发生。
These hallmarks include primary ageing mechanisms, namely disabled macroautophagy, loss of proteostasis, genomic instability, and epigenetic alterations as well as additional downstream aberrations, including mitochondrial dysfunction, dysregulated neurohormonal signalling, cell senescence and inflammation. Beyond mechanistic and heuristic considerations, we also emphasize the question of how each of these hallmarks can be therapeutically targeted to attenuate residual cardiovascular risk and extend the healthspan.
这些特征包括主要的衰老机制,即巨自噬功能障碍、蛋白稳态丧失、基因组不稳定性和表观遗传改变,以及其他下游畸变,包括线粒体功能障碍、神经激素信号失调、细胞衰老和炎症。 除了机制性和启发性考虑,我们还强调如何针对这些特征进行治疗,以减轻残余心血管风险并延长健康寿命。
Hallmark 1: disabled macroautophagy
标志 1:巨自噬功能障碍
Macroautophagy, hereafter referred to as autophagy, is a key cellular quality control mechanism that is essential for the clearance (and subsequent replacement) of dysfunctional cytoplasmic components. During autophagy, potentially toxic protein aggregates and ageing organelles are sequestrated in double-membraned vesicles, the autophagosomes, which subsequently fuse with lysosomes for enzymatic degradation of their cargo, thereby providing metabolites for bioenergetic and anabolic reactions23. As such, autophagy not only contributes to proteostasis but also targets other macromolecules, such as ectopic DNA in the cytosol, lipid droplets (lipophagy), portions of the endoplasmic reticulum (reticulophagy), entire organelles, including mitochondria (mitophagy) and peroxisomes (pexophagy), as well as invading pathogens (xenophagy), through highly specialized subroutines. Furthermore, the autophagic machinery contributes to ‘heterophagy’24, a process by which cardiomyocytes package dysfunctional mitochondria in so-called exospheres to extrude the waste material into the extracellular space, where it is degraded by cardiac-resident macrophages25. In view of its vital homeostatic role, autophagy is widely accepted to govern the healthspan and lifespan of various eukaryotic cell types, particularly those that comprise the cardiovascular system26,27,28. Indeed, dysfunctional autophagy is implicated in a broad range of cardiovascular disorders, including atherosclerosis, coronary heart disease, diabetic cardiomyopathy, chemotherapy-induced cardiotoxicity, arrhythmia and heart failure27,29,30. Moreover, several widely used cardioprotective drugs, including aspirin, β-adrenergic blockers and calcium-channel blockers, have been shown to regulate autophagic activity31,32,33,34.
巨自噬,以下简称自噬,是一种关键的细胞质量控制机制,对清除(以及随后的替换)功能失调的细胞质成分至关重要。 在自噬过程中,潜在的毒性蛋白聚集物和衰老的细胞器被隔离在双层膜囊泡(即自噬体)中,自噬体随后与溶酶体融合,对其所载物质进行酶降解,从而为生物能量和合成代谢反应提供代谢物 23 。 因此,自噬不仅有助于蛋白质稳态,还能通过高度专业化的子程序靶向其他大分子,如胞浆中的异位 DNA、脂滴(脂噬)、内质网部分区域(内质网噬)、包括线粒体(线粒体噬)和过氧化物酶体(过氧化物酶体噬)在内的整个细胞器,以及入侵的病原体(异物噬)。 此外,自噬机制还有助于“异噬” 24 ,通过这个过程,心肌细胞将功能失调的线粒体包裹在所谓的胞外体中,将废物排出到细胞外空间,在那里它们被心脏驻留巨噬细胞降解 25 。鉴于其重要的稳态作用,自噬被广泛认为可以调控各种真核细胞类型的健康寿命和寿命,特别是那些构成心血管系统的细胞 26,27,28 。 事实上,功能障碍的自噬与多种心血管疾病有关,包括动脉粥样硬化、冠心病、糖尿病心肌病、化疗引起的心脏毒性、心律失常和心力衰竭 27,29,30 。此外,几种广泛使用的心脏保护药物,包括阿司匹林、β-肾上腺素能阻滞剂和钙通道阻滞剂,已被证明可以调节自噬活性 31,32,33,34 。
Mounting evidence supports the notion that autophagy declines with organismal ageing35,36, thereby contributing to the pathogenesis of age-related chronic diseases37. In the cardiovascular system, a progressive reduction in autophagy has been reported in the vasculature of both aged mice and humans38. By contrast, whether and how ageing affects autophagy in the heart has been a matter of debate39. Indeed, the abundance of autophagosomes in a tissue does not inform on autophagic flux (because autophagosomes result from both the sequestration of autophagic cargo and their failed disposal by lysosomal hydrolases), and specific quantification protocols that capture the dynamics of the process are required40,41. Therefore, refined approaches to evaluate autophagic flux have been used in studies of flies, mice and rats, providing irrefutable evidence that autophagy declines in the ageing heart as well as in the ageing vasculature42,43,44,45.
越来越多的证据表明,自噬会随着机体衰老而下降 35,36 ,从而促进与年龄相关的慢性疾病的发生 37 。在心血管系统中,有报道称衰老的小鼠和人类的血管中自噬逐渐减少 38 。相比之下,衰老是否以及如何影响心脏中的自噬一直备受争议 39 。 的确,组织中自噬体的丰富程度并不能说明自噬通量(因为自噬体是由于自噬货物的隔离以及溶酶体水解酶对其处理失败而产生的),因此需要能够捕捉该过程动态的特定量化方案 40,41 。 因此,在果蝇、小鼠和大鼠的研究中,已经使用了改进的方法来评估自噬通量,提供了无可辩驳的证据,表明自噬在衰老的心脏以及衰老的血管中都会下降 42,43,44,45 。
Several mechanisms are involved in the age-related decline of autophagy. Among these mechanisms, the inhibitory hyperacetylation of autophagy proteins46 has been associated with reduced activity of sirtuin deacetylases and their obligatory substrate NAD+ in the aged cardiovascular system47. Inhibition of autophagy might also be explained by the increased provision of nutrients, which provide excessive acetyl CoA levels for protein acetylation, and overactivation of the insulin–IGF1–serine/threonine-protein kinase mTOR signalling pathway, especially in the presence of (visceral) obesity and metabolic syndrome. Calcium signalling, which is altered during cardiovascular ageing48, has also been proposed to regulate autophagy, at least in endothelial cells and cardiomyocytes49,50. In addition, an age-associated reduction in levels of the polyamine spermidine51,52 might compromise the hypusination-dependent translation of autophagy proteins53. Indeed, cardiac spermidine content progressively declines with age in humans (Fig. 3), and its exogenous supplementation reactivates both cardiac and vascular autophagy in aged mice54,55.
有几种机制参与了自噬的年龄相关性下降。在这些机制中,自噬蛋白的抑制性过度乙酰化 46 与衰老的心血管系统中 sirtuin 去乙酰化酶及其必需底物 NAD + 的活性降低有关 47 。 自噬抑制也可能通过营养物质的增加来解释,营养物质为蛋白质乙酰化提供了过量的乙酰辅酶 A 水平,并过度激活了胰岛素-IGF1-丝氨酸/苏氨酸蛋白激酶 mTOR 信号通路,尤其是在存在(内脏)肥胖和代谢综合征的情况下。钙信号在心血管衰老过程中会发生改变 48 ,据称也可以调节自噬,至少在内皮细胞和心肌细胞中是这样 49,50 。 此外,与年龄相关的亚精胺水平降低 51,52 可能会损害依赖次牛基化作用的自噬蛋白翻译 53 。事实上,人类心脏中的亚精胺含量随着年龄的增长而逐渐下降(图 3),而外源性补充亚精胺可以重新激活老年小鼠的心脏和血管自噬 54,55 。
图 3:人类心脏中亚精胺水平随年龄下降。

The cardiac concentration of spermidine, which regulates many cellular processes, declines with ageing. Spermidine concentrations were measured in left ventricular samples from human donors and explanted failing hearts from men and women (n = 35). Pearson correlation coefficient (r) and P value are indicated, together with the corresponding linear regression line (solid line) and 95% confidence intervals (dashed lines). Procedures involving these samples were approved by the Ethical Committee of the Medical University of Graz (28-508 ex 15/16) and were conducted in accordance with the principles outlined in the Declaration of Helsinki.
亚精胺调节许多细胞过程,其心脏浓度随年龄增长而下降。在来自男性和女性的人类捐献者和移植的衰竭心脏的左心室样本中测量亚精胺浓度(n = 35)。图中显示了 Pearson 相关系数(r)和 P 值,以及相应的线性回归线(实线)和 95%置信区间(虚线)。 涉及这些样本的程序已获得格拉茨医科大学伦理委员会的批准(28-508 ex 15/16),并按照《赫尔辛基宣言》中概述的原则进行。
Notably, genetic suppression of autophagy in cardiomyocytes, vascular smooth muscle cells or endothelial cells phenocopies aspects of cardiovascular ageing (Table 1). For instance, mice harbouring a cardiomyocyte-specific deficiency of the autophagy gene Atg5 have disrupted sarcomere structure, impaired excitation–contraction coupling, dysfunctional mitochondria and increased oxidative stress56,57. These alterations are coupled to left ventricular dysfunction, accelerated cardiac hypertrophy, reduced effort tolerance and prematurely compromised cardiopulmonary function56,57. In vascular smooth muscle cells, knockout of Atg7 impairs calcium mobilization and contractility, accelerates senescence, and exacerbates diet-induced atherogenesis58,59. Even endothelial cells, which have a comparatively high proliferative potential and can be routinely replaced, might be affected by defective autophagy. For instance, despite no obvious abnormalities at rest, mice with vascular endothelial-specific Atg5 and Atg7 ablation show a defect in haemostasis due to impaired synthesis and secretion of von Willebrand factor60. Similarly, endothelium-specific knockout of the gene encoding transcriptional activator of autophagy Krüppel-like factor 4 (Klf4) causes early-onset endothelial dysfunction as denoted by reduced acetylcholine-induced vasodilatation in vivo61. Collectively, autophagy impairments affecting various cell types in the cardiovascular system entail structural and functional abnormalities reminiscent of cardiovascular ageing.
值得注意的是,心肌细胞、血管平滑肌细胞或内皮细胞中自噬的基因抑制会模拟心血管衰老的某些方面(表 1)。例如,携带心肌细胞特异性自噬基因 Atg5 缺陷的小鼠具有紊乱的肌节结构、受损的兴奋-收缩耦联、功能障碍的线粒体和增加的氧化应激 56,57 。 这些改变与左心室功能障碍、加速的心脏肥大、降低的运动耐量和过早受损的心肺功能有关 56,57 。在血管平滑肌细胞中,敲除 Atg7 会损害钙的动员和收缩能力,加速衰老,并加剧饮食诱导的动脉粥样硬化 58,59 。 即使是内皮细胞,其具有相对较高的增殖能力并且可以常规地被替换,也可能受到缺陷自噬的影响。例如,尽管在静息状态下没有明显的异常,但血管内皮特异性 Atg5 和 Atg7 敲除的小鼠由于血管性血友病因子合成和分泌受损而表现出血止血功能缺陷 60 。 类似地,内皮细胞特异性敲除自噬转录激活因子 Krüppel 样因子 4 (Klf4) 的编码基因会导致早发性内皮功能障碍,表现为体内乙酰胆碱诱导的血管舒张减少 61 。总而言之,影响心血管系统中各种细胞类型的自噬损伤会导致让人联想到心血管衰老的结构和功能异常。
表 1 通过基因手段加强心血管衰老标志物的啮齿动物模型
Conversely, genetic, dietary or pharmacological interventions resulting in autophagy activation effectively delay cardiovascular ageing and extend lifespan26 (Table 2). For instance, ubiquitous activation of autophagy in mice by a knock-in mutation in the gene encoding beclin 1 (Becn1F121A/F121A) attenuates age-related cardiac abnormalities, including myocardial fibrosis, hypertrophy and apoptosis, coupled to lifespan extension62. Similarly, endothelium-specific activation of autophagy by a Klp4 transgene attenuates age-related vascular dysfunction61. Caloric restriction, the most robust autophagy-inducing intervention, also delays typical features of cardiovascular ageing, such as diastolic dysfunction, cardiac fibrosis and hypertrophy, high blood pressure, vascular stiffness, and systemic inflammation63,64,65. Similarly, in mice and rats, the natural polyamine and autophagy inducer spermidine elicits cardiac and vascular geroprotective effects such as attenuated myocardial thickening, stiffness and diastolic dysfunction, improved vascular endothelial dysfunction, blood pressure regulation, arterial stiffening, ventricular–vascular coupling and reduced chronic low-grade inflammation51,54,55. The effects of spermidine seem to be largely dependent on autophagy given that Atg5-deficient mice show no cardioprotection after spermidine supplementation55. Other pro-autophagic compounds66, such as rapamycin and trehalose, also exert anti-ageing effects on the heart and the vasculature, supporting the notion that disabled autophagy is an actionable hallmark of cardiovascular ageing38,67,68,69,70 (Table 2).
相反,通过遗传、饮食或药物干预激活自噬可以有效延缓心血管衰老并延长寿命 26 (表 2)。例如,在小鼠中,通过敲入编码 beclin 1 的基因(Becn1 F121A/F121A )中的突变来普遍激活自噬,可以减轻与年龄相关的心脏异常,包括心肌纤维化、肥大和细胞凋亡,并延长寿命 62 。 同样地,Klp4 转基因对内皮细胞自噬的特异性激活可以减轻与年龄相关的血管功能障碍 61 。热量限制是最有效的自噬诱导干预措施,它也能延缓典型的心血管衰老特征,如舒张功能障碍、心肌纤维化和肥大、高血压、血管僵硬和全身炎症 63,64,65 。 同样地,在小鼠和大鼠中,天然多胺和自噬诱导剂亚精胺会引发心脏和血管的抗衰老效应,例如减轻心肌增厚、僵硬和舒张功能障碍,改善血管内皮功能障碍、血压调节、动脉僵硬、心室-血管耦合以及减少慢性低度炎症 51,54,55 。 亚精胺的作用似乎很大程度上取决于自噬,因为缺乏 Atg5 的小鼠在补充亚精胺后没有表现出心脏保护作用 55 。其他促进自噬的化合物 66 ,如雷帕霉素和海藻糖,也对心脏和血管产生抗衰老作用,这支持了自噬功能障碍是心血管衰老的可操作标志的观点 38,67,68,69,70 (表 2)。
表 2 通过作用于衰老特征来延长心血管健康寿命的干预措施
Hallmark 2: loss of proteostasis
标志 2:蛋白稳态丧失
Proteostasis is a major driver of organismal health and stress resistance71. The delicate balance between protein synthesis, folding and degradation is tightly regulated by an extensive network of molecular chaperones and proteolytic enzymes72. The integrity of the proteome is maintained by refolding denatured proteins or their selective degradation. Therefore, when heat shock proteins (HSPs) and other molecular chaperones fail to refold a protein into its correct conformation, the dysfunctional protein must be destroyed by the ubiquitin–proteasome system (UPS) or chaperone-mediated autophagy (CMA). CMA is a selective form of protein degradation, in which heat shock cognate 71 kDa protein binds to the KFERQ motif of specific proteins designated for degradation, facilitating their translocation into the lysosome via the receptor lysosome-associated membrane glycoprotein 2A (LAMP2A)73. The efficiency of this extensive and resource-demanding proteostasis network declines with age, leading to the accumulation of misfolded and damaged proteins that often collate to form non-functional protein aggregates74. Although the formation of potentially toxic protein aggregates is most prominent in neurodegenerative disorders, it is also evident in a wide range of age-related cardiovascular diseases, including atherosclerosis, atrial fibrillation and heart failure as well as hypertrophic, ischaemic and dilated cardiomyopathies75,76,77,78.
蛋白质稳态是驱动机体健康和抗压性的主要因素 71 。蛋白质合成、折叠和降解之间的微妙平衡受到由分子伴侣和蛋白水解酶组成的广泛网络的严格调控 72 。蛋白质组的完整性通过使变性蛋白质重新折叠或选择性降解来维持。 因此,当热休克蛋白(HSP)和其他分子伴侣未能将蛋白质重新折叠成正确的构象时,功能失调的蛋白质必须通过泛素-蛋白酶体系统(UPS)或伴侣介导的自噬(CMA)进行破坏。 CMA 是一种选择性的蛋白质降解形式,其中热休克同源 71 kDa 蛋白与特定蛋白质的 KFERQ 基序结合,这些蛋白质被指定降解,从而促进它们通过受体溶酶体相关膜糖蛋白 2A (LAMP2A)转运到溶酶体 73 。 随着年龄增长,这种广泛且消耗大量资源的蛋白质稳态网络效率下降,导致错误折叠和受损蛋白质的积累,这些蛋白质通常会聚集形成无功能的蛋白质聚集体 74 。 虽然潜在的毒性蛋白聚集体的形成在神经退行性疾病中最为突出,但在各种与年龄相关的心血管疾病中也很明显,包括动脉粥样硬化、心房颤动和心力衰竭,以及肥厚型、缺血性和扩张型心肌病 75,76,77,78 。
The age-related loss of proteostasis poses a particular challenge for cardiomyocytes79,80 because their functional units — sarcomeres — rely on proteostasis for maintenance81. However, cardiomyocytes face an extraordinary load of proteotoxic reactive oxygen species (ROS) that are generated as a by-product of intense oxidative phosphorylation. Aged rat hearts have been shown to accumulate oxidized and ubiquitinated proteins, correlating with reduced proteasome activity and expression levels of several HSPs82,83. Similarly, the hearts of aged mice have an elevated level of protein ubiquitination84, coinciding with a major accumulation of protein aggregates85. In the vasculature, CMA apparently declines with age as indicated by low expression of the genes encoding CMA-related proteins and low CMA activity scores in the aortas of aged but otherwise healthy individuals86. The single-cell human transcriptomic atlas (Tabula Sapiens) indicates that the expression of CMA-relevant genes diminishes with age, mostly in aortic macrophages and smooth muscle cells86,87. An age-related loss of proteostasis is further denoted by augmented ubiquitin levels and impaired proteasome activity in carotid plaques from older individuals (mean age 72 years)88. Similarly, aged rat aortas have reduced basal expression and stress-induced activation of HSP70 (ref. 89), and aortic tissues from old mice and almost all humans aged >50 years accumulate medin amyloid aggregates90,91. Notably, vascular medin aggregates correlate with cerebrovascular dysfunction in aged mice and cognitive decline in patients with vascular dementia or Alzheimer disease90,92,93.
与年龄相关的蛋白稳态丧失对心肌细胞 79,80 提出了特殊的挑战,因为它们的功能单元——肌小节——依赖于蛋白稳态进行维护 81 。然而,心肌细胞面临着巨大的蛋白毒性活性氧(ROS)负荷,这些活性氧是剧烈的氧化磷酸化反应的副产物。 衰老的大鼠心脏已被证明会积累氧化和泛素化的蛋白质,这与蛋白酶体活性降低和几种热休克蛋白 (HSP) 的表达水平相关 82,83 。 类似地,衰老小鼠的心脏具有较高水平的蛋白质泛素化 84 ,这与蛋白质聚集体的大量积累相一致 85 。 在血管系统中,CMA 显然会随着年龄的增长而下降,这体现在老年但其他方面健康的个体的主动脉中,编码 CMA 相关蛋白的基因表达量低,CMA 活性评分也较低 86 。单细胞人类转录组图谱(Tabula Sapiens)表明,CMA 相关基因的表达随着年龄的增长而减少,主要是在主动脉巨噬细胞和平滑肌细胞中 86,87 。 与年龄相关的蛋白质稳态丧失进一步表现为老年人(平均年龄 72 岁)的颈动脉斑块中泛素水平升高和蛋白酶体活性受损 88 。同样,老年大鼠的主动脉 HSP70 的基底表达和应激诱导的激活均降低(参考文献 89 ),并且老年小鼠的主动脉组织和几乎所有年龄 >50 岁的人类都会积累髓心蛋白淀粉样蛋白聚集体 90,91 。 值得注意的是,血管髓质蛋白聚集与老年小鼠的脑血管功能障碍以及血管性痴呆或阿尔茨海默病患者的认知能力下降有关 90,92,93 。
The causal role of protein instability in cardiovascular ageing is supported by experimental studies in which cardiovascular ageing is accentuated (or delayed) by inducing (or preventing) the collapse of proteostasis in the cardiovascular system (Table 1). For instance, a missense mutation in the molecular chaperone and small HSP αB-crystallin (CryAB), which binds to misfolded proteins to prevent their aggregation, causes desmin mislocalization to protein aggregates, cardiac remodelling and heart failure in young mice94,95,96, recapitulating human desmin-related cardiomyopathy97. In accordance with these findings, accumulation of cardiac desmin occurs in progeroid Lmna–/– mice, thereby compromising cardiac conduction and causing progressive dilated cardiomyopathy98,99, which can be rescued in vivo by cardiac-specific overexpression of Cryab100 or in vitro by SUMO-conjugating enzyme UBC9-induced activation of the UPS101. Another example of cardiac proteotoxicity is provided by mice expressing a heart-specific transgene encoding an aggregate-prone protein with 83 glutamine repeats (PQ83), which causes cardiac dysfunction and dilatation and, subsequently, death102. Similarly, knockdown of E2 ubiquitin-conjugating enzyme compromises the UPS and causes premature cardiac ageing and dysfunction in flies103. By contrast, pharmaceutical induction of HSP70 using geranylgeranylacetone reduces myocardial protein aggregates and improves cardiac function and survival in Cryab-mutant mice104. Geranylgeranylacetone also ameliorates functional recovery from cardiac ischaemia–reperfusion injury in wild-type rats105. Similarly, stabilizing the protein transthyretin using the chaperone-mimicking drug tafamidis improves quality of life and reduces cardiovascular hospitalizations and mortality in patients with a late-onset form of amyloid cardiomyopathy that mainly affects men106,107. In the vasculature, blockade of CMA in LAMP2A-deficient mice promotes atherosclerotic plaque formation by favouring dyslipidaemia, vascular smooth muscle cell dedifferentiation and a pro-inflammatory macrophage phenotype86. Conversely, systemic activation of CMA in mice overexpressing human LAMP2A attenuates the severity and slows the progression of atherosclerosis induced by a proatherogenic diet86. In aged mice, genetic ablation of the amyloid medin precursor, lactadherin (also called milk fat globule-Egf factor 8 and encoded by Mfge8), reverses age-related cerebrovascular dysfunction90. Notably, the beneficial effects of caloric restriction in slowing cardiovascular ageing are also linked to improved proteostasis108 as suggested by the upregulation of HSP70 and several transcripts involved in protein folding in cardiac and skeletal muscles of rodents and humans, respectively83,109. Taken together, convergent data implicate proteostasis collapse in age-related cardiovascular decline and suggest that genetic, dietary or pharmacological strategies that maintain proteostasis can delay cardiovascular ageing (Table 2).
蛋白质不稳定性在心血管衰老中的因果作用得到了实验研究的支持,在这些研究中,通过诱导(或阻止)心血管系统中蛋白稳态的崩溃,心血管衰老被加剧(或延缓)(表 1)。 例如,分子伴侣和小 HSP αB-晶体蛋白(CryAB)中的一个错义突变,该蛋白与错误折叠的蛋白质结合以防止其聚集,会导致结蛋白错误定位到蛋白质聚集体、年轻小鼠的心脏重塑和心力衰竭 94,95,96 ,重现人类结蛋白相关的心肌病 97 。 与这些发现一致,早衰型 Lmna –/– 小鼠体内会出现心脏结蛋白的积累,从而损害心脏传导,导致进行性扩张型心肌病 98,99 ,而这可以通过心脏特异性过表达 Cryab 100 在体内挽救,或通过 SUMO 结合酶 UBC9 诱导的 UPS 激活 101 在体外挽救。 心脏蛋白毒性的另一个例子是表达心脏特异性转基因的小鼠,该转基因编码具有 83 个谷氨酰胺重复序列(PQ83)的易聚集蛋白,这会导致心脏功能障碍和扩张,并最终导致死亡 102 。类似地,敲低 E2 泛素结合酶会损害 UPS,并导致果蝇过早的心脏老化和功能障碍 103 。 相比之下,使用香叶基香叶丙酮通过药物诱导 HSP70,可以减少 Cryab 突变小鼠的心肌蛋白聚集,并改善心脏功能和存活率 104 。香叶基香叶丙酮还能改善野生型大鼠从心脏缺血再灌注损伤中的功能恢复 105 。 同样地,使用伴侣蛋白模拟药物氯苯唑酸稳定转甲状腺素蛋白,可以提高主要影响男性的迟发性淀粉样变心肌病患者的生活质量,并减少心血管住院和死亡率 106,107 。在血管系统中,LAMP2A 缺陷小鼠的 CMA 阻断通过促进血脂异常、血管平滑肌细胞去分化和促炎巨噬细胞表型,促进动脉粥样硬化斑块的形成 86 。 相反,在过度表达人 LAMP2A 的小鼠中,CMA 的全身激活可以减轻由促动脉粥样硬化饮食诱导的动脉粥样硬化的严重程度,并延缓其进展 86 。在老年小鼠中,淀粉样蛋白 medin 前体,乳凝集素(也称为乳脂球-Egf 因子 8,由 Mfge8 编码)的基因消融可以逆转与年龄相关的脑血管功能障碍 90 。 值得注意的是,热量限制在延缓心血管衰老方面的有益作用也与改善蛋白质稳态有关 108 ,这由啮齿动物和人类心肌和骨骼肌中 HSP70 的表达上调以及参与蛋白质折叠的多个转录本所证实 83,109 。 总而言之,大量趋同的数据表明蛋白质稳态崩溃与年龄相关的心血管功能衰退有关,并表明维持蛋白质稳态的遗传、饮食或药物策略可以延缓心血管衰老(表 2)。
Hallmark 3: genomic instability
标志 3:基因组不稳定性
Although genomic instability is a common feature of ageing in various organs and across species, the rate at which mutations accumulate varies substantially between tissues110. In the cardiovascular system, age-related mutations seem to be largely dependent on both tissue and context. The murine heart shows substantial genomic and transcriptomic stability, at least until the age of 2 years111,112, a time point at which the decline in cardiac function and structure becomes evident at the cellular (Box 2) and whole-organ (Box 1) levels4,26. As such, although mutations in the heart in the form of genome rearrangements were reported in transgenic (LacZ) mice, especially at an older age (33 months)112,113, these alterations are probably not a cause but rather a consequence of other hallmarks of cardiac ageing. Therefore, unlike more proliferative organs (such as the liver and small intestine)111, the heart might be more resilient to age-related DNA mutations. Along similar lines, whether the chromosomes of aged human and mouse hearts undergo telomere shortening after early postnatal life stages114,115 is increasingly contested115,116,117. Indeed, even though genetically induced telomere attrition causes cardiac senescence (Table 1) and hyperlong telomeres reduce cardiovascular risk in mice118, telomere shortening-associated cardiac disease in humans119,120 seems to be largely independent of age119.
尽管基因组不稳定性是不同器官和物种衰老的常见特征,但突变的累积速率在不同组织之间差异很大 110 。在心血管系统中,与年龄相关的突变似乎主要取决于组织和环境。 至少在 2 岁之前,小鼠的心脏显示出相当大的基因组和转录组稳定性 111,112 ,但此时细胞(方框 2)和整个器官(方框 1)水平上的心脏功能和结构下降变得明显 4,26 。 因此,尽管在转基因 (LacZ) 小鼠的心脏中发现了基因组重排形式的突变,尤其是在老年(33 个月)时 112,113 ,但这些改变可能不是心脏老化的其他标志的原因,而是结果。因此,与更多增殖性器官(如肝脏和小肠) 111 不同,心脏可能更能抵抗与年龄相关的 DNA 突变。 与此类似,衰老的人类和鼠类心脏的染色体是否在出生后早期阶段发生端粒缩短 114,115 ,正受到越来越多的质疑 115,116,117 。事实上,即使基因诱导的端粒损耗会导致心脏衰老(表 1),而超长端粒可以降低小鼠的心血管风险 118 ,但人类与端粒缩短相关的心脏疾病 119,120 似乎在很大程度上与年龄无关 119 。
The relationship between DNA stability and vascular ageing is less controversial121, although ageing seems to cause greater DNA damage and telomere dysfunction in vascular endothelial cells than in smooth muscle cells122. DNA damage has also been reported in human atherosclerotic plaques123,124, and mice with vascular smooth muscle cell-restricted DNA damage show lower atherosclerotic plaque stability despite the negligible effect on plaque size124. Furthermore, DNA damage has been linked to features of accelerated ageing in the carotid and cerebral arteries in human and mouse radiation studies, thereby implicating telomere attrition and cell senescence as potential mechanisms125,126,127. Genetic and, therefore, more specific disruption of DNA integrity in mutant mice also induces a phenotype reminiscent of vascular ageing. For instance, defective nucleotide excision repair in Ercc1Δ/– mice causes endothelial dysfunction, increased vascular stiffness and elevated blood pressure at a very young age (8–16 weeks)128. This phenotype is partially recapitulated when the mutation in Ercc1 is restricted to vascular endothelial or smooth muscle cells129,130. Similarly, mutant mice with low levels of the spindle assembly checkpoint protein BUBR1 (also known as mitotic checkpoint serine/threonine-protein kinase BUB1β) have increased arterial stiffness, reduced arterial elasticity and impaired vasorelaxation by the age of 3–5 months131. Notably, aged wild-type mice have low aortic expression of BUBR1 (ref. 131). Comprehensive and systematic genome analyses are needed to evaluate vascular DNA damage during the life course of naturally aged mice and humans to corroborate the relevance of these findings. These studies will also help to pinpoint the exact role of genomic instability in vascular ageing as radiation-induced genetic models of forced DNA damage show both systemic abnormalities and other mechanisms that might promote vascular dysfunction, irrespective of DNA integrity status.
DNA 稳定性与血管衰老之间的关系争议较小 121 ,尽管衰老似乎会导致血管内皮细胞的 DNA 损伤和端粒功能障碍比在平滑肌细胞中更严重 122 。人类动脉粥样硬化斑块中也已报告存在 DNA 损伤 123,124 ,并且血管平滑肌细胞限制性 DNA 损伤的小鼠表现出较低的动脉粥样硬化斑块稳定性,尽管对斑块大小的影响可以忽略不计 124 。 此外,DNA 损伤与人和小鼠辐射研究中颈动脉和脑动脉的加速衰老特征有关,从而暗示端粒损耗和细胞衰老是潜在的机制 125,126,127 。在突变小鼠中,基因及因此更特异性的 DNA 完整性破坏也会诱导类似于血管衰老的表型。 例如,在 Ercc1 Δ/– 小鼠中,有缺陷的核苷酸切除修复会导致内皮功能障碍、血管僵硬度增加以及在非常年轻时(8-16 周)血压升高 128 。当 Ercc1 的突变仅限于血管内皮细胞或平滑肌细胞时,这种表型会被部分重现 129,130 。 同样地,纺锤体组装检查点蛋白 BUBR1(也称为有丝分裂检查点丝氨酸/苏氨酸蛋白激酶 BUB1β)低水平的突变小鼠,在 3-5 个月大时,动脉僵硬度增加,动脉弹性降低,血管舒张受损 131 。值得注意的是,年老的野生型小鼠主动脉 BUBR1 的表达量较低(参考文献 131 )。 为了证实这些发现的相关性,需要对自然衰老的小鼠和人类生命历程中的血管 DNA 损伤进行全面和系统的基因组分析。这些研究还有助于确定基因组不稳定性在血管衰老中的确切作用,因为辐射诱导的强制 DNA 损伤遗传模型显示出全身性异常和其他可能促进血管功能障碍的机制,而与 DNA 完整性状态无关。
Notably, the age-related mutations with the highest prevalence and effect on the cardiovascular system do not occur in the heart or vasculature but instead in the blood and bone marrow. Depending on detection thresholds132, 10–40% of individuals aged 70–79 years are estimated to carry somatic mutations in their peripheral blood cells133,134. Remarkably, a substantial proportion (~20% on average) of peripheral blood cells in these individuals might be derived from a single mutated stem cell132,134. Although the carriers of these mutations are at risk of haematological malignancies134,135, the majority are aged but otherwise healthy individuals. Therefore, the term clonal haematopoiesis of indeterminate potential (CHIP; that is, the presence of a somatic mutation in the blood of individuals who have no major haematological abnormalities) was coined to describe this condition136. Key among CHIP somatic mutations are those affecting the genes encoding the transcriptional regulators DNA (cytosine-5)-methyltransferase 3A (DNMT3A), methylcytosine dioxygenase TET2 (TET2), polycomb group protein ASXL1 (ASXL1) and tyrosine-protein kinase JAK2 (JAK2)134,135,137. Epidemiological studies have revealed that carriers of these mutations are at increased risk of coronary heart disease, coronary artery calcification, myocardial infarction and ischaemic heart failure138,139,140. Indeed, the risk of cardiovascular diseases associated with CHIP is similar to that conferred by traditional risk factors, including hypertension, smoking and elevated plasma total cholesterol level132. Furthermore, growing evidence indicates that the cardiovascular risk associated with central obesity and metabolic syndrome is associated with increased bone marrow haematopoiesis and high circulating leukocyte counts, which might exacerbate CHIP as well as immune cell accumulation in the heart and vessels, thereby fuelling chronic inflammation141,142.
值得注意的是,对心血管系统影响最大、且与年龄相关的突变并非发生在心脏或血管中,而是在血液和骨髓中。据估计,根据检测阈值 132 ,年龄在 70-79 岁的人群中,有 10-40%的人其外周血细胞携带体细胞突变 133,134 。值得注意的是,在这些人中,很大一部分(平均约 20%)外周血细胞可能来源于单个突变的干细胞 132,134 。 尽管这些突变的携带者有患血液系统恶性肿瘤的风险 134,135 ,但大多数是年长的但其他方面健康的个体。因此,创造了“意义未明的克隆性造血”(CHIP)一词(即,在没有重大血液学异常的个体的血液中存在体细胞突变)来描述这种情况 136 。 CHIP 体细胞突变中,最关键的是那些影响转录调节因子的基因,包括 DNA(胞嘧啶-5)-甲基转移酶 3A(DNMT3A)、甲基胞嘧啶双加氧酶 TET2(TET2)、多梳蛋白组蛋白 ASXL1(ASXL1)和酪氨酸蛋白激酶 JAK2(JAK2) 134,135,137 。流行病学研究表明,这些突变的携带者患冠心病、冠状动脉钙化、心肌梗死和缺血性心力衰竭的风险增加 138,139,140 。 事实上,与传统风险因素(包括高血压、吸烟和血浆总胆固醇水平升高) 132 相关的 CHIP 的心血管疾病风险与之相似。 此外,越来越多的证据表明,与中心型肥胖和代谢综合征相关的心血管风险与骨髓造血功能增强和高循环白细胞计数有关,这可能会加剧 CHIP 以及免疫细胞在心脏和血管中的积累,从而加剧慢性炎症 141,142 。
A causal role of CHIP in cardiovascular disease is inferred from experimental studies showing that CHIP somatic mutations promote atherosclerosis, pathological cardiac remodelling and dysfunction, and heart failure in mouse models138,143,144,145,146. Mechanistically, the cardiovascular adverse effects of CHIP are linked to inflammation as both human and mouse carriers of CHIP somatic mutations have a pro-inflammatory phenotype in their immune cells138,143,144,145,147. By contrast, the increased cardiovascular risk in individuals with CHIP is almost completely abolished in the absence of functional IL-6 signalling148, and suppression of the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome–IL-1β axis protects against atherosclerosis and cardiac dysfunction in mice with Tet2-deficient bone marrow143,144. Notably, although TET2 levels decline in the myeloid cells of aged mice and humans, thereby promoting pro-inflammatory tumour necrosis factor (TNF) signalling149, Tet2–/– mice show no signs of accelerated cardiac ageing150. These mice spontaneously develop a phenotype that is reminiscent of human pulmonary arterial hypertension by the age of 7–10 months, which regresses in response to anti-inflammatory treatment with an antibody targeting IL-1β150.
CHIP 在心血管疾病中的因果作用推断来自实验研究,这些研究表明,CHIP 体细胞突变会促进小鼠模型中的动脉粥样硬化、病理性心脏重塑和功能障碍以及心力衰竭 138,143,144,145,146 。从机制上讲,CHIP 对心血管的不良影响与炎症有关,因为 CHIP 体细胞突变的人类和老鼠携带者在其免疫细胞中都具有促炎表型 138,143,144,145,147 。 相比之下,在没有功能性 IL-6 信号的情况下,CHIP 个体心血管风险的增加几乎完全消失 148 ,并且抑制 NACHT、LRR 和 PYD 结构域蛋白 3(NLRP3)炎性体–IL-1β轴可以保护具有 Tet2 缺陷骨髓的小鼠免受动脉粥样硬化和心脏功能障碍 143,144 。 值得注意的是,尽管 TET2 水平在年老的小鼠和人类的髓系细胞中下降,从而促进促炎性肿瘤坏死因子(TNF)信号传导 149 ,但 Tet2 –/– 小鼠没有表现出加速心脏衰老的迹象 150 。这些小鼠在 7-10 个月大时自发地出现了一种类似于人类肺动脉高压的表型,这种表型可以通过使用靶向 IL-1β的抗体进行抗炎治疗来消退 150 。
Taken together, CHIP somatic mutations might contribute to the age-related deterioration of cardiovascular health. Targeting these mutated clones or their downstream pro-inflammatory signalling might be a valid strategy to attenuate residual cardiovascular risk in older individuals (discussed further in section Hallmark 8). However, whether the genomic instability of cells that compose the cardiovascular structures themselves constitutes a target for anti-ageing interventions is not yet clear.
总而言之,CHIP 体细胞突变可能导致与年龄相关的心血管健康恶化。靶向这些突变克隆或其下游促炎信号传导可能是减弱老年人残留心血管风险的有效策略(在第 8 个标志中进一步讨论)。然而,构成心血管结构的细胞自身的基因组不稳定性是否构成抗衰老干预的目标尚不清楚。
Hallmark 4: epigenetic alterations
标志 4:表观遗传改变
Independent of DNA mutations, gene expression levels in the cardiovascular system might be affected by a variety of epigenetic factors, including DNA methylation, histone modifications and non-coding RNAs (ncRNAs)151,152. These epigenetic alterations converge on age-related shifts and imbalances in gene transcription with links to pathophysiological processes such as loss of proteostasis, mitochondrial dysfunction and inflammation153,154. Even in the heart, where genomic instability is contested (see Hallmark 3), advancing age causes pronounced heterogeneity in gene expression155, suggesting that epigenetic alterations might be involved in cardiovascular ageing.
独立于 DNA 突变,心血管系统中的基因表达水平可能受到多种表观遗传因素的影响,包括 DNA 甲基化、组蛋白修饰和非编码 RNA (ncRNA) 151,152 。这些表观遗传改变集中于与年龄相关的基因转录变化和失衡,与病理生理过程有关,例如蛋白质稳态丧失、线粒体功能障碍和炎症 153,154 。 即使在心脏中,基因组不稳定性备受争议(见标志 3),但年龄的增长会导致基因表达的显著异质性 155 ,这表明表观遗传改变可能与心血管衰老有关。
Non-coding RNAs 非编码 RNA
Since their discovery, ncRNAs have emerged as highly relevant epigenetic factors in cardiovascular medicine156. Indeed, various subfamilies of ncRNAs, including circular RNAs, microRNAs (miRNAs) and long ncRNAs (lncRNAs), have been linked to senescence and disabled autophagy, among other hallmarks of cardiovascular ageing. For instance, circFOXO3, a circular RNA generated from the gene encoding the forkhead box protein O3 transcription factor (Foxo3 or FOXO3), is highly expressed in hearts from aged mice and humans157. In a mouse model of doxorubicin-induced cardiomyopathy, a plasmid encoding circFOXO3 promoted cardiac senescence and exacerbated contractile function, whereas small interfering RNA targeting endogenous circFOXO3 had senolytic and cardioprotective effects157.
自从 ncRNA 被发现以来,已经成为心血管医学中高度相关的表观遗传因素 156 。实际上,包括环状 RNA、microRNA (miRNA) 和长链 ncRNA (lncRNA) 在内的多个 ncRNA 亚家族都与衰老和自噬功能障碍有关,以及其他心血管衰老的标志。 例如,circFOXO3 是一种由编码叉头框蛋白 O3 转录因子(Foxo3 或 FOXO3)的基因生成的环状 RNA,在老年小鼠和人类的心脏中高表达 157 。在阿霉素诱导的心肌病小鼠模型中,编码 circFOXO3 的质粒促进了心脏衰老并加剧了收缩功能,而靶向内源性 circFOXO3 的小干扰 RNA 则具有衰老抑制和心脏保护作用 157 。
The cardiac expression profile of several miRNAs changes with age158. A noticeable example is miRNA-34a, which is highly expressed in the myocardium of aged mice and humans159, and actually promotes senescence and DNA damage159,160. By contrast, aged mice with miRNA-34a knockout have a rejuvenated cardiac phenotype, characterized by reduced cardiomyocyte hypertrophy and apoptosis as well as by improved cardiac function159. Notably, miRNA-34a is also upregulated in aged mouse aortas161, and inhibition of miRNA-34a renders vascular smooth muscle cells less prone to senescence in vitro162, while improving cardiac recovery from coronary artery occlusion in vivo159. miRNA-34a-knockout mice also have reduced vascular calcification induced by toxic doses of vitamin D162. Mice and humans also have an age-dependent increase in the cardiac expression level of miRNA-22, a ncRNA with pro-hypertrophic and autophagy-inhibitory effects163,164. Accordingly, miRNA-22 inhibition promotes autophagy in cardiac cells in vitro, while reducing structural and functional decline after myocardial infarction in vivo, particularly in older mice164.
几种 miRNA 的心脏表达谱随年龄而变化 158 。一个显著的例子是 miRNA-34a,它在老年小鼠和人类的心肌中高表达 159 ,并且实际上促进衰老和 DNA 损伤 159,160 。相比之下,miRNA-34a 敲除的老年小鼠具有恢复活力的心脏表型,其特征是心肌细胞肥大和凋亡减少,以及心脏功能得到改善 159 。 值得注意的是,miRNA-34a 在老年小鼠的主动脉中也表达上调 161 ,抑制 miRNA-34a 可使血管平滑肌细胞在体外不易衰老 162 ,同时改善体内冠状动脉闭塞后的心脏恢复 159 。miRNA-34a 敲除小鼠的血管钙化也减少,而血管钙化是由毒性剂量的维生素 D 诱导的 162 。 小鼠和人类的心脏 miRNA-22 表达水平也会随着年龄的增长而增加,miRNA-22 是一种具有促进肥大和抑制自噬作用的非编码 RNA 163,164 。因此,抑制 miRNA-22 可以促进体外培养的心脏细胞的自噬,同时减少体内(尤其是在老年小鼠中)心肌梗死后的结构和功能衰退 164 。
Despite the ongoing debate as to whether lncRNAs are important transcriptional regulators or a manifestation of transcriptional noise165,166, the lncRNA Sarrah was found to be downregulated in aged mouse hearts as well as in rodent models of ischaemic cardiomyopathy and heart failure with preserved ejection fraction (HFpEF)167. Conversely, overexpression of Sarrah improves cardiomyocyte survival, endothelial cell proliferation and functional recovery from myocardial ischaemia, at least in young mice167. Overexpression of Sarrah also reduces cardiac apoptosis in aged mice167; however, whether this effect provides functional benefits in the aged heart or vasculature remains to be determined.
尽管关于 lncRNA 是重要的转录调节因子还是转录噪音的表现,目前仍存在争议 165,166 ,但研究发现,lncRNA Sarrah 在老年小鼠心脏以及缺血性心肌病和射血分数保留型心力衰竭(HFpEF)的啮齿动物模型中均有下调 167 。 相反,至少在幼鼠中,Sarrah 的过度表达可以提高心肌细胞的存活率、内皮细胞的增殖以及心肌缺血后的功能恢复 167 。Sarrah 的过度表达还可以减少老年小鼠的心脏细胞凋亡 167 ;然而,这种效应是否能为老年心脏或血管带来功能上的益处仍有待确定。
Histone modifications 组蛋白修饰
Post-translational modifications of histones have a major role in epigenetic regulation. For instance, acetylation of histone lysine residues modulates gene transcription activity by altering chromatin structure condensation. In this regard, cardiac and vascular expression levels of NAD+-dependent protein deacetylase sirtuin 1 (SIRT1) were shown to decline with advancing age in mice and also in human failing hearts168,169. Importantly, moderate restoration of SIRT1 expression in mice delays cardiac ageing as indicated by enhanced cardiac stress resistance and reduced markers of senescence and age-dependent adverse myocardial remodelling170. Similarly, transgenic mice overexpressing Sirt1 specifically in the endothelium have reduced age-related decline in vasodilatory responses171. By contrast, global or endothelium-specific ablation of Sirt6, encoding another nuclear histone deacetylase, causes early-onset vascular dysfunction172, and reduced SIRT6 expression in vascular smooth muscle cells is linked to an increased risk of vascular calcification in mice and humans with chronic kidney disease173. SIRT6-deficient mice also develop a progeroid cardiac phenotype, denoted by myocardial hypertrophic and degenerative alterations, whereas SIRT6 overexpression protects against pathological cardiac hypertrophy induced by transverse aortic constriction or isoprenaline infusion174.
组蛋白的翻译后修饰在表观遗传调控中起着重要作用。例如,组蛋白赖氨酸残基的乙酰化通过改变染色质结构的凝聚来调节基因转录活性。在这方面,NAD + 依赖性蛋白脱乙酰酶 sirtuin 1 (SIRT1) 的心脏和血管表达水平在小鼠体内以及人类衰竭心脏中均显示出随着年龄的增长而下降 168,169 。 重要的是,在小鼠中适度恢复 SIRT1 的表达可以延缓心脏衰老,这表现在增强的心脏抗压能力,以及减少的衰老标志物和年龄依赖性的不良心肌重塑 170 。类似地,在内皮细胞中特异性过表达 Sirt1 的转基因小鼠,其血管舒张反应的年龄相关性下降有所减少 171 。 相比之下,Sirt6(编码另一种核组蛋白去乙酰化酶)的全身或内皮细胞特异性消融会导致早发性血管功能障碍 172 ,而血管平滑肌细胞中 SIRT6 表达的降低与小鼠和患有慢性肾病的人类血管钙化风险增加有关 173 。 SIRT6 缺陷型小鼠也会出现早衰型心脏表型,表现为心肌肥大和退行性改变,而 SIRT6 过表达可防止横主动脉缩窄或异丙肾上腺素输注引起的心脏病理性肥大 174 。
Genetic ablation of genes encoding the histone deacetylase (HDAC) family members HDAC1, HDAC2, HDAC3, HDAC5, HDAC7 or HDAC9 revealed the importance of histone (de)acetylation for the development, maturation and growth of cardiomyocytes175. Pharmacological inhibition of HDACs improves diastolic dysfunction — the key sign of cardiac ageing — in old mice as well as in murine and feline models of premature diastolic impairment176,177,178. However, further efforts are warranted to determine whether and to what extent altered histone acetylation mediates the cardiac benefits of pan-HDAC inhibitors. This question is particularly important because gain-of-function and loss-of-function mutations of specific HDACs have revealed unique, and sometimes opposing, roles of the various HDAC isoforms in cardiovascular homeostasis and pathology179. For instance, HDAC6-deficient mice, which have reduced susceptibility to pulmonary hypertension and stress-induced systolic dysfunction180,181, manifest premature onset of diastolic dysfunction that progresses with ageing182. Similarly, knockout of Hdac9 protects against vascular calcification183 but accentuates pathological hypertrophy in relevant mouse models184. Therefore, selective targeting of HDAC isoforms, ideally in a cell type-specific manner, might be an effective strategy for reducing adverse and off-target effects.
对编码组蛋白去乙酰化酶 (HDAC) 家族成员 HDAC1、HDAC2、HDAC3、HDAC5、HDAC7 或 HDAC9 的基因进行基因消融,揭示了组蛋白(去)乙酰化对心肌细胞发育、成熟和生长的重要性 175 。HDAC 的药物抑制作用改善了老年小鼠以及小鼠和猫的早发性舒张功能障碍模型中的舒张功能障碍——心脏衰老的关键标志 176,177,178 。 然而,还需要进一步的努力来确定组蛋白乙酰化修饰的改变是否以及在多大程度上介导了泛-HDAC 抑制剂对心脏的益处。这个问题尤其重要,因为特定 HDAC 的功能获得性突变和功能丧失性突变揭示了各种 HDAC 亚型在心血管稳态和病理学中独特且有时相反的作用 179 。 例如,HDAC6 缺陷小鼠对肺动脉高压和应激诱导的收缩功能障碍的易感性降低 180,181 ,但表现出随年龄增长而进展的舒张功能障碍的早发 182 。类似地,Hdac9 的敲除可以防止血管钙化 183 ,但会加剧相关小鼠模型中的病理性肥大 184 。 因此,有选择性地靶向 HDAC 亚型(最好是以细胞类型特异性的方式)可能是减少不良反应和脱靶效应的有效策略。
Numerous studies have shown that histone methylation has an important role in cardiac homeostasis at least in early life185,186,187,188. Indeed, targeting histone methylation has been proposed as a potential therapeutic approach for treating atherosclerosis and hypertrophic cardiomyopathy on the basis of preclinical experiments188,189,190. Nonetheless, further research is required to determine whether natural ageing is accompanied by altered and targetable histone methylation in the cardiovascular system.
大量研究表明,组蛋白甲基化在心脏稳态中具有重要作用,至少在生命早期是这样 185,186,187,188 。事实上,基于临床前实验,靶向组蛋白甲基化已被提议作为治疗动脉粥样硬化和肥厚型心肌病的潜在治疗方法 188,189,190 。然而,还需要进一步的研究来确定自然衰老是否伴随着心血管系统中组蛋白甲基化的改变和可靶向性。
DNA methylation DNA 甲基化
Although profiling DNA methylation patterns is increasingly used to estimate the rate of cardiovascular ageing191,192,193, the contribution of altered DNA methylation to cardiovascular disease remains controversial. For instance, although the cardiac DNA methylome is altered in human failing hearts194,195,196, measurements of DNA methylation in human cardiac samples underestimate chronological age by a decade compared with those performed on blood and other tissues197,198. This finding suggests that the human heart might be comparatively resistant to age-related alterations in DNA methylation. By contrast, a rat study revealed an age-dependent decline in the cardiac expression level of DNA (cytosine-5)-methyltransferase 1 (DNMT1), which is involved in the maintenance of DNA methylation199. However, cardiac Dnmt1 knockout in adult mouse hearts did not cause any obvious maladaptive consequences, at least until the age of 12 months, and unexpectedly had cardioprotective effects in a model of cardiotoxicity199. The effect of other DNA methyltransferases, such as DNMT3A and DNMT3B, which are required for de novo DNA methylation, also remains controversial. Although the levels of DNMT3A and DNMT3B are upregulated in human heart failure196, discordant reports from studies of mice with conditional cardiomyocyte-specific deletion of Dnmt3a and/or Dnmt3b showed either no detrimental phenotype or severe cardiomyopathy200,201. Therefore, the effect of modulating cardiac DNA methylation during adulthood, let alone in ageing, is perplexing, even though the embryonic ablation of DNA methyltransferases is known to cause premature death due to cardiac anomalies, among other developmental defects202.
尽管分析 DNA 甲基化模式越来越多地用于评估心血管衰老的速度 191,192,193 ,但 DNA 甲基化改变对心血管疾病的贡献仍然存在争议。例如,虽然人类衰竭心脏的心脏 DNA 甲基化组发生了改变 194,195,196 ,但与在血液和其他组织上进行的测量相比,人类心脏样本中 DNA 甲基化的测量低估了十年时间 197,198 。 这一发现表明,与年龄相关的 DNA 甲基化改变相比,人类心脏可能具有较强的抵抗力。相比之下,一项大鼠研究显示,参与维持 DNA 甲基化的 DNA(胞嘧啶-5)-甲基转移酶 1(DNMT1)的心脏表达水平随年龄增长而下降 199 。 然而,在成年小鼠心脏中敲除心脏 Dnmt1 并没有引起任何明显的适应不良后果,至少在 12 个月大之前没有,并且出乎意料地在心脏毒性模型中具有心脏保护作用 199 。其他 DNA 甲基转移酶(如 DNMT3A 和 DNMT3B,它们是 de novo DNA 甲基化所必需的)的作用仍然存在争议。 尽管 DNMT3A 和 DNMT3B 的水平在人类心力衰竭中上调 196 ,但来自条件性心肌细胞特异性敲除 Dnmt3a 和/或 Dnmt3b 的小鼠研究报告不一致,显示要么没有不利的表型,要么是严重的心肌病 200,201 。 因此,在成年期(更不用说在衰老期)调节心脏 DNA 甲基化的效果令人费解,即使已知胚胎消融 DNA 甲基转移酶会导致早夭,原因是心脏畸形以及其他发育缺陷 202 。
In terms of vascular DNA methylation, opposing observations have also been reported in aged versus atherosclerotic vessels. Despite the intimate association between ageing and atherosclerosis203, ageing is associated with global vascular DNA hypomethylation204, whereas atherosclerotic vessels seem to show focal DNA hypermethylation205, and atherosclerotic lesions can even be attenuated by pharmacological inhibition of DNMT1 (ref. 206). Therefore, further investigation is required to elucidate the importance and mechanistic role of reduced vascular DNA methylation in natural ageing.
就血管 DNA 甲基化而言,在衰老血管与动脉粥样硬化血管中也观察到了相反的现象。尽管衰老与动脉粥样硬化之间存在密切联系 203 ,但衰老与整体血管 DNA 低甲基化有关 204 ,而动脉粥样硬化血管似乎表现出局部 DNA 高甲基化 205 ,甚至可以通过药物抑制 DNMT1 来减轻动脉粥样硬化病变(参考文献 206 )。 因此,需要进一步研究来阐明血管 DNA 甲基化降低在自然衰老中的重要性和机制作用。
Hallmark 5: mitochondrial dysfunction
标志 5:线粒体功能障碍
Mitochondrial dysfunction is a key characteristic of ageing and age-related chronic diseases207,208, particularly in the cardiovascular system, which heavily relies on mitochondrial metabolism and signalling. Beyond their function in myocardial bioenergetics, the mitochondria have a crucial role in intracellular calcium homeostasis, redox balance, anabolic and catabolic reactions, and in the initiation of inflammatory and lethal signals in almost all cell types in the cardiovascular system209,210,211. Therefore, age-related mitochondrial defects preferentially affect the cardiovascular system212,213. Indeed, mitochondrial dysfunction is arguably one of the most crucial aetiological factors across a wide range of cardiovascular disorders214,215.
线粒体功能障碍是衰老和与年龄相关的慢性疾病的一个关键特征 207,208 ,尤其是在心血管系统中,心血管系统严重依赖线粒体代谢和信号传导。除了在心肌生物能量学中的功能外,线粒体在细胞内钙稳态、氧化还原平衡、合成代谢和分解代谢反应,以及在心血管系统中几乎所有细胞类型的炎症和致命信号的启动中都起着至关重要的作用 209,210,211 。 因此,与年龄相关的线粒体缺陷优先影响心血管系统 212,213 。事实上,线粒体功能障碍可以说是各种心血管疾病最重要的病因因素之一 214,215 。
In the heart, ageing is associated with a sizable decline in mitochondrial content55,216. In addition, aged cardiac mitochondria are structurally and functionally compromised as indicated by an increase in diameter (swelling), matrix deformation, loss of cristae and reduced oxidative phosphorylation secondary to reduced activity and stability of mitochondrial respiratory chain complexes, particularly complex IV55,213,217. Although these abnormalities might mainly affect interfibrillar mitochondria (that is, those located between myofibrils)212,218,219, cardiac metabolism is substantially impaired. Therefore, aged human and mouse hearts are characterized by limited metabolic flexibility and increased reliance on glycolysis rather than fatty acid oxidation220,221.
在心脏中,衰老与线粒体含量的大幅下降有关 55,216 。此外,衰老的心脏线粒体在结构和功能上都会受到损害,表现为直径增加(肿胀)、基质变形、嵴丢失以及氧化磷酸化降低,而这继发于线粒体呼吸链复合物(尤其是复合物 IV)的活性和稳定性降低 55,213,217 。 尽管这些异常可能主要影响肌纤维间线粒体(即位于肌原纤维之间的线粒体) 212,218,219 ,但心脏代谢会受到严重损害。因此,衰老的人类和小鼠心脏的特征是代谢灵活性受限,并且更加依赖糖酵解而非脂肪酸氧化 220,221 。
Mitochondrial defects are also evident in the aged vasculature5,6 given that both vascular endothelial and smooth muscle cells have impaired mitochondrial biogenesis, coupled to reduced mitochondrial mass and respiration222,223. Vascular ageing is also associated with exaggerated mitochondrial production of ROS, which directly scavenge and reduce the bioavailability of nitric oxide, thereby impairing endothelium-dependent vasorelaxation224,225. In addition, suppressed antioxidative activity of the transcriptional regulator nuclear factor erythroid 2-related factor 2 (NRF2)226,227 renders aged vascular cells particularly susceptible to ROS-induced damage. Indeed, NRF2-deficient mice have an accelerated vascular ageing phenotype coupled with increased susceptibility to vascular senescence and cerebromicrovascular dysfunction228,229. Regardless, excessive production of mitochondrial ROS might increase the rate of mitochondrial DNA (mtDNA) mutations and deletions and even cause the release of mtDNA from mitochondria, thereby activating pro-inflammatory signalling pathways. Therefore, ROS lead to the activation of nuclear factor-κB (NF-κB), whereas cytosolic mtDNA triggers the DNA sensor cyclic GMP–AMP synthase (cGAS) and subsequently the cyclic GMP–AMP receptor stimulator of interferon genes protein (STING) to initiate a type I interferon response6,230. These pro-inflammatory pathways drive age-related endothelial dysfunction231,232,233. Supporting this scenario, aged vessels show a positive feedback loop between mitochondrial dysfunction and IL-6, which exacerbates hyperlipidaemia-induced atherogenesis in mice234. Another relevant feature explaining specific facets of mitochondrial dysfunction is the decline in levels of key metabolic cofactors, particularly NAD+, which is likely to contribute to cardiac and vascular ageing235,236.
线粒体缺陷在衰老的血管系统中也很明显 5,6 ,因为血管内皮细胞和平滑肌细胞的线粒体生物合成受损,同时线粒体质量和呼吸作用下降 222,223 。血管衰老还与线粒体过度产生 ROS 有关,ROS 直接清除并降低一氧化氮的生物利用度,从而损害内皮依赖性血管舒张 224,225 。 此外,转录调节因子核因子 E2 相关因子 2 (NRF2) 226,227 的抗氧化活性受到抑制,使得衰老的血管细胞特别容易受到 ROS 诱导的损伤。事实上,缺乏 NRF2 的小鼠表现出血管加速衰老的表型,同时更容易发生血管衰老和脑微血管功能障碍 228,229 。 无论如何,线粒体活性氧的过度产生可能会增加线粒体 DNA(mtDNA)突变和缺失的速率,甚至导致 mtDNA 从线粒体释放,从而激活促炎信号通路。 因此,活性氧会导致核因子-κB(NF-κB)的激活,而胞质 mtDNA 会触发 DNA 传感器环磷酸鸟苷-腺苷酸合成酶(cGAS),并随后触发环磷酸鸟苷-腺苷酸受体干扰素基因刺激蛋白(STING),从而启动 I 型干扰素反应 6,230 。这些促炎通路会驱动与年龄相关的内皮功能障碍 231,232,233 。 为了支持这一观点,衰老血管显示出线粒体功能障碍和 IL-6 之间的正反馈回路,这加剧了小鼠体内高脂血症诱导的动脉粥样硬化 234 。解释线粒体功能障碍某些方面的另一个相关特征是关键代谢辅因子,特别是 NAD+水平的下降 + ,这可能导致心脏和血管衰老 235,236 。
Mechanistically, reduced mitochondrial quality control due to age-related loss of mitophagy might explain the accumulation of dysfunctional organelles and the associated decline in cardiovascular homeostasis28,237,238. Supporting this notion, mice deficient in mitochondrial serine/threonine-protein kinase PINK1 (also known as PTEN-induced putative kinase protein 1), which have increased age-dependent accumulation of dysfunctional mitochondria due to impaired mitophagy239,240, develop premature cardiac hypertrophy and systolic dysfunction at an early age241. Similarly, increasing mitochondrial biogenesis by overexpression of peroxisome proliferator-activated receptor-γ coactivator-α (PGC1A) specifically in cardiomyocytes accelerates cardiac ageing and shortens lifespan, despite initially improving cardiac health242. The accentuated cardiac ageing phenotype in these transgenic mice was attributed, at least in part, to increased accumulation of damaged mitochondria due to failed quality control of newly generated organelles by an overwhelmed mitophagy machinery242. Along similar lines, abrogating mitochondrial dynamics (that is, fusion and fission) in cardiomyocytes substantially impairs mitophagy, which coincides with accelerated mitochondrial senescence and premature signs of cardiac ageing in Mfn1–Mfn2–Drp1 triple knockout mice243. Similarly, defective endothelial mitophagy in Prkaa-knockout mice is associated with mitochondrial fragmentation and vascular endothelial dysfunction, which can be rescued by Atg7 overexpression or in vivo administration of the mTOR-dependent autophagy inducer rapamycin244. By contrast, the cardioprotective effects of spermidine in aged mice and rats are linked to improved mitochondrial structure and function through induction of mitophagy51,55,219.
从机制上讲,线粒体质量控制因年龄相关的线粒体自噬丧失而降低,这可能解释了功能失调的细胞器的积累以及相关的心血管稳态下降 28,237,238 。 支持这一观点的是,缺乏线粒体丝氨酸/苏氨酸蛋白激酶 PINK1(也称为 PTEN 诱导的假定激酶蛋白 1)的小鼠,由于线粒体自噬受损导致功能失调的线粒体随年龄增长而积累增加 239,240 ,会在早期出现早发性心脏肥大和收缩功能障碍 241 。 类似地,通过在心肌细胞中特异性地过度表达过氧化物酶体增殖物激活受体-γ辅激活因子-α (PGC1A) 来增加线粒体生物合成会加速心脏衰老并缩短寿命,尽管最初改善了心脏健康 242 。 在这些转基因小鼠中,心脏衰老表型的加剧至少部分归因于受损线粒体的积累增加,这是由于不堪重负的线粒体自噬机制未能对新产生的细胞器进行质量控制 242 。 与此类似,在心肌细胞中消除线粒体动力学(即融合和分裂)会大大损害线粒体自噬,这与 Mfn1–Mfn2–Drp1 三重敲除小鼠中线粒体衰老加速和心脏衰老早期迹象同时发生 243 。 类似地,Prkaa 敲除小鼠中缺陷的内皮线粒体自噬与线粒体碎片化和血管内皮功能障碍有关,而这些可以通过 Atg7 过表达或体内施用 mTOR 依赖性自噬诱导剂雷帕霉素来挽救 244 。相比之下,亚精胺在老年小鼠和大鼠中的心脏保护作用与通过诱导线粒体自噬来改善线粒体结构和功能有关 51,55,219 。
Systemic disruption of mitochondrial homeostasis precipitates the age-associated deterioration of cardiovascular health in mouse models245. For instance, a knock-in mutation in the nucleus-encoded catalytic subunit of polymerase-γ (Polga), rendering this mtDNA polymerase proofreading deficient, leads to excessive mtDNA point mutations and deletions as well as to increased ROS production246,247. These mtDNA-mutator mice have a shortened lifespan and show accelerated myocardial senescence, fibrosis, hypertrophy and dilatation as well as left ventricular systolic and diastolic dysfunction246,247. Moreover, these mice manifest premature vascular stiffening, reduced vascular compliance and increased atherosclerosis223,248. Notably, overexpressing the antioxidant enzyme catalase or treatment with the mitochondria-targeted antioxidant SkQ1 attenuate aspects of accelerated cardiovascular ageing in mtDNA-mutator mice246,249. Another example of mitochondrial defects driving cardiac ageing is provided by mice lacking either of mitochondrial pyruvate carrier subunits 1 or 2 in cardiomyocytes, which causes cardiac hypertrophy, abnormal contractility and premature death250,251. Even vascular endothelial cells, which largely rely on glycolysis rather than oxidative phosphorylation for energy production252, lose ex vivo control of vascular tone after pharmacological inhibition of respiratory chain complexes or the mitochondrial pyruvate carrier253. Moreover, in vivo disruption of endothelial fatty acid oxidation by conditional deletion of mitochondrial carnitine O-palmitoyltransferase 2 promotes valvular disease and increases vascular permeability in several organs254. Therefore, forced induction of mitochondrial dysfunction accelerates cardiovascular ageing and increases susceptibility to cardiovascular disease at a young age.
在线粒体稳态中的全身性紊乱会加速小鼠模型中与年龄相关的心血管健康恶化 245 。例如,在细胞核编码的聚合酶-γ(Polga)催化亚基中的敲入突变,导致这种 mtDNA 聚合酶缺乏校对能力,从而导致过度的 mtDNA 点突变和缺失,以及 ROS 产生增加 246,247 。 这些 mtDNA 突变小鼠的寿命缩短,并表现出加速的心肌衰老、纤维化、肥大和扩张,以及左心室收缩和舒张功能障碍 246,247 。此外,这些小鼠还表现出过早的血管僵硬、血管顺应性降低和动脉粥样硬化增加 223,248 。 值得注意的是,过表达抗氧化酶过氧化氢酶或使用靶向线粒体的抗氧化剂 SkQ1 可以减轻 mtDNA 突变小鼠加速心血管衰老的某些方面 246,249 。线粒体缺陷驱动心脏衰老的另一个例子是心肌细胞中缺少线粒体丙酮酸载体亚基 1 或 2 的小鼠,这会导致心脏肥大、收缩异常和过早死亡 250,251 。 即使是血管内皮细胞,它们主要依靠糖酵解而非氧化磷酸化来产生能量 252 ,在药理学抑制呼吸链复合物或线粒体丙酮酸载体后,也会丧失对血管张力的离体控制 253 。 此外,通过条件性删除线粒体肉碱 O-棕榈酰转移酶 2 来破坏体内内皮细胞脂肪酸氧化,会促进瓣膜疾病并增加多个器官的血管通透性 254 。因此,强制诱导线粒体功能障碍会加速心血管衰老,并增加在年轻时患心血管疾病的易感性。
Importantly, interventions designed to avoid certain aspects of age-related mitochondrial dysfunction have shown cardiovascular protective effects and delayed ageing. For instance, mitophagy induced in cardiomyocytes by transgenic overexpression of E3 ubiquitin-protein ligase parkin not only maintains mitochondrial integrity and cardiac function but also attenuates signs of senescence and inflammation in aged mice238. Mitochondria-targeted antioxidant agents, such as mitoquinone mesylate (MitoQ; Antipodean Pharmaceuticals) and SS-31 (elamipretide), normalize vascular oxidative stress and restore the vasodilatory function of endothelial cells in aged mice and humans255,256. Consistent with these findings, overexpression of the antioxidant catalase in mitochondria delays cardiac ageing and extends the lifespan of mice257. Furthermore, reducing mtDNA damage in apolipoprotein E-knockout (Apoe−/−) mice by overexpressing Twinkle mtDNA helicase improves mitochondrial function and the stability of atherosclerotic plaques by increasing the thickness of fibrous caps and reducing the formation of necrotic cores258. Similarly, boosting mitochondrial oxidative metabolism by supplementation with NAD+ precursors exerts cytoprotective effects on mouse and human cells in vitro259, and delays both cardiac and vascular signs of ageing in vivo260,261. Therefore, nicotinamide can suppress age-related cardiac hypertrophy and diastolic dysfunction in mice260. Moreover, in aged mice, administration of nicotinamide mononucleotide reduces age-related vascular and (cerebro)microvascular endothelial dysfunction, avoids arterial stiffness by preserving elastin content and decreasing collagen accumulation, and increases cerebral and skeletal muscle blood flow, thereby improving neurovascular coupling, cognitive performance and exercise endurance capacity261,262,263,264. Restoring NAD+ levels by inhibiting its consuming enzymes also recapitulates some of these cardiovascular benefits235,265. Taken together, these findings indicate that mitochondria are an actionable target to prevent or delay cardiovascular ageing at the preclinical level, calling for attempts to translate these findings to the clinic215.
重要的是,旨在避免与年龄相关的线粒体功能障碍某些方面干预措施已经显示出心血管保护作用并延缓衰老。例如,转基因过度表达 E3 泛素-蛋白连接酶 parkin 在心肌细胞中诱导的线粒体自噬不仅维持线粒体完整性和心脏功能,而且减轻了老年小鼠的衰老和炎症迹象 238 。 靶向线粒体的抗氧化剂,如甲磺酸美托醌(MitoQ;Antipodean Pharmaceuticals)和 SS-31(elamipretide),可以使衰老小鼠和人体内的血管氧化应激正常化,并恢复内皮细胞的血管舒张功能 255,256 。与这些发现一致的是,在线粒体中过度表达抗氧化剂过氧化氢酶可以延缓心脏衰老,并延长小鼠的寿命 257 。 此外,通过过表达 Twinkle mtDNA 解旋酶来减少载脂蛋白 E 敲除 (Apoe −/− ) 小鼠的 mtDNA 损伤,可以通过增加纤维帽的厚度和减少坏死核的形成来改善线粒体功能和动脉粥样硬化斑块的稳定性 258 。 同样地,通过补充 NAD + 前体来促进线粒体氧化代谢,对小鼠和人类细胞在体外具有细胞保护作用 259 ,并延缓了体内心脏和血管衰老的迹象 260,261 。因此,烟酰胺可以抑制小鼠体内与年龄相关的心脏肥大和舒张功能障碍 260 。 此外,在老年小鼠中,给予烟酰胺单核苷酸可减少与年龄相关的血管和(脑)微血管内皮功能障碍,通过保持弹性蛋白含量和减少胶原积累来避免动脉僵硬,并增加大脑和骨骼肌的血流量,从而改善神经血管耦合、认知能力和运动耐力 261,262,263,264 。 通过抑制消耗 NAD + 的酶来恢复 NAD 水平也能重现部分心血管益处 235,265 。综上所述,这些发现表明线粒体是一个可操作的靶点,可以在临床前水平预防或延缓心血管衰老,因此需要尝试将这些发现转化为临床应用 215 。
Hallmark 6: cell senescence
标志 6:细胞衰老
Senescence is generally defined as permanent arrest of the cell cycle, leading to irreversible loss of replicative capacity coupled with a reduction in specific cellular functions and the acquisition of pro-inflammatory features. However, senescence does not apply only to proliferative cell types, such as endothelial cells and fibroblasts, but can also affect cardiomyocytes117,266,267. Senescence is induced by several internal and external stressors, including telomere shortening, oncogenic signalling, persistent DNA damage, oxidative and mechanical stress, nutrient imbalance and mitochondrial dysfunction as well as viral or bacterial infections268,269. Senescent cells secrete a range of pro-inflammatory cytokines and chemokines, growth factors, and matrix proteases — collectively referred to as the senescence-associated secretory phenotype (SASP) — which can trigger other non-senescent cells to undergo ‘secondary’ (also known as ‘contagious’ or ‘paracrine’) senescence270. However, the identification of senescent cells is challenging, especially in vivo, due to the absence of specific biomarkers, and relies on a combination of parameters, including the activity of lysosomal senescence-associated β-galactosidase and the expression of cyclin-dependent kinase inhibitors such as CDKN2A (p16) and CDKN1A (p21)271,272. Notably, even though cell senescence is required for normal development, wound healing and protection against neoplastic transformation, a growing body of evidence suggests that uncontrolled accumulation of senescent cells (that is, without sufficient elimination by immune cells)273 might contribute to organismal ageing and related chronic diseases266,274. Indeed, cellular senescence has been implicated in multiple age-related cardiovascular diseases, including atherosclerosis, ischaemic cardiomyopathy and HFpEF275,276,277,278,279. The accumulation of senescent cells has been shown to blunt the regenerative potential of aged skeletal muscle280 but whether this process also occurs in the myocardium remains to be demonstrated.
衰老通常被定义为细胞周期的永久性停滞,导致复制能力的不可逆丧失,同时伴随着特定细胞功能的下降和促炎性特征的获得。然而,衰老不仅适用于增殖性细胞类型,如内皮细胞和成纤维细胞,而且还会影响心肌细胞 117,266,267 。 衰老是由多种内在和外在的压力源诱导的,包括端粒缩短、癌基因信号、持续性 DNA 损伤、氧化和机械应力、营养失衡和线粒体功能障碍以及病毒或细菌感染 268,269 。 衰老细胞会分泌一系列促炎性细胞因子和趋化因子、生长因子和基质蛋白酶,这些统称为衰老相关分泌表型 (SASP),它们可以触发其他非衰老细胞发生“继发性”(也称为“传染性”或“旁分泌性”)衰老 270 。 然而,由于缺乏特异性生物标志物,衰老细胞的识别具有挑战性,尤其是在体内,并且依赖于多种参数的组合,包括溶酶体衰老相关β-半乳糖苷酶的活性以及细胞周期蛋白依赖性激酶抑制剂(如 CDKN2A (p16) 和 CDKN1A (p21))的表达 271,272 。 值得注意的是,即使细胞衰老是正常发育、伤口愈合和防止肿瘤形成所必需的,但越来越多的证据表明,衰老细胞的过度积累(即免疫细胞清除不足) 273 可能导致机体衰老和相关的慢性疾病 266,274 。 事实上,细胞衰老与多种年龄相关的心血管疾病有关,包括动脉粥样硬化、缺血性心肌病和射血分数保留型心力衰竭 275,276,277,278,279 。研究表明,衰老细胞的积累会削弱老年骨骼肌的再生潜力 280 ,但这一过程是否也发生在心肌中仍有待证实。
Owing to their anatomical location, vascular endothelial cells (including those in the cerebral microcirculation) are directly exposed to senescence-inducing stimuli, including biochemical and haemodynamic factors281,282,283. Compared with their normal counterparts, senescent endothelial cells show abnormal morphology and enlargement as well as increased adhesion to the basement membrane and reduced capacity to align with the direction of blood flow281,284. Functionally, senescent endothelial cells have been implicated in the age-related incapacity of endothelium-dependent vasodilatation285 as well as in the loss of blood–brain barrier integrity286. Similarly, senescent cardiomyocytes have a maladaptive phenotype, characterized by hypertrophy and profibrotic signalling, which might contribute to typical features of cardiac ageing, including myocardial wall thickening and stiffness (Box 1), respectively117,287. Reduction of telomere length coupled with excessive cell cycling cannot account for cardiomyocyte senescence; however, telomere damage without shortening could be induced by mitochondrial dysfunction and oxidative stress117. In accordance with their role in wound healing288, senescent fibroblasts restricted myocardial fibrosis in a mouse model of transverse aortic constriction289. However, whether this phenomenon is also relevant in age-related myocardial fibrosis is unknown and raises the possibility that senescence in different cell types has context-dependent, positive and negative roles in cardiovascular physiology. Indeed, specific subtypes of senescent cells might perform different physiological roles more generally (for example, wound healing, limitation of sterile inflammation and the maintenance of the hepatic blood–tissue barrier) and their elimination could result in mixed outcomes282,290.
由于其解剖位置,血管内皮细胞(包括脑微循环中的内皮细胞)直接暴露于诱导衰老的刺激,包括生化和血流动力学因素 281,282,283 。与正常的内皮细胞相比,衰老的内皮细胞表现出异常的形态和增大,以及对基底膜的粘附力增加和与血流方向对齐的能力降低 281,284 。 在功能上,衰老的内皮细胞与年龄相关的内皮依赖性血管舒张功能丧失 285 以及血脑屏障完整性的丧失 286 有关。类似地,衰老的心肌细胞具有适应不良的表型,其特征在于肥大和促纤维化信号传导,这可能导致心脏衰老的典型特征,包括心肌壁增厚和僵硬(框 1) 117,287 。 端粒长度的缩短加上过度的细胞周期不能解释心肌细胞的衰老;然而,没有缩短的端粒损伤可能是由线粒体功能障碍和氧化应激诱导的 117 。与它们在伤口愈合中的作用相一致 288 ,衰老的成纤维细胞限制了横向主动脉缩窄小鼠模型中的心肌纤维化 289 。 然而,这种现象是否也与年龄相关的心肌纤维化有关尚不清楚,并提出了不同细胞类型的衰老在心血管生理学中具有依赖于环境的正反两方面作用的可能性。 事实上,特定亚型的衰老细胞可能在更广泛的范围内发挥不同的生理作用(例如,伤口愈合、限制无菌性炎症以及维持肝脏血-组织屏障),而清除它们可能会导致好坏参半的结果 282,290 。
Nevertheless, in naturally aged mice, systemic clearance of senescent cells can delay ageing and extend lifespan287,291,292. In the cardiovascular system of these animals, genetic or pharmacological elimination of senescent cells attenuates myocardial hypertrophy and fibrosis and maintains cardiac function117. Clearance of cells expressing p16Ink4a was shown to improve cardiac stress resistance and attenuate myocardial remodelling in response to β-adrenergic stimulation of aged mice287. Similarly, long-term senolytic therapy delays vascular ageing as indicated by improved endothelium-dependent vasodilatation293, and also improves neurovascular coupling and cognitive function294. Conversely, accelerated senescence induced by telomere uncapping or telomere shortening causes an early-onset cardiovascular ageing phenotype characterized by premature myocardial hypertrophy, fibrosis and diastolic dysfunction as well as vascular oxidative stress, endothelial dysfunction and elevated blood pressure in mice242,285,295,296. This finding might also be relevant to humans because increased vascular expression of senescence biomarkers is associated with vascular endothelial dysfunction in sedentary older individuals297. Moreover, the effects of cardiac glycosides298 might be partially attributable to their senolytic activity299.
然而,在自然衰老的小鼠中,全身清除衰老细胞可以延缓衰老并延长寿命 287,291,292 。在这些动物的心血管系统中,通过基因或药物手段清除衰老细胞可以减轻心肌肥大和纤维化,并维持心脏功能 117 。研究表明,清除表达 p16 的细胞 Ink4a 可以提高老年小鼠的心脏抗应激能力,并减轻β-肾上腺素刺激引起的心肌重塑 287 。 同样地,长期使用衰老细胞清除剂可以延缓血管衰老,表现为内皮依赖性血管舒张的改善 293 ,并且还可以改善神经血管耦合和认知功能 294 。 相反,由端粒解封或端粒缩短引起的加速衰老会导致早发性心血管衰老表型,其特征是小鼠出现过早的心肌肥大、纤维化和舒张功能障碍,以及血管氧化应激、内皮功能障碍和血压升高 242,285,295,296 。 这一发现可能也与人类相关,因为久坐不动的老年人血管内衰老生物标志物表达的增加与血管内皮功能障碍有关 297 。此外,强心苷 298 的作用可能部分归因于其衰老细胞清除活性 299 。
Taken together, these findings suggest that accumulating secretory and metabolically active senescent cells in the cardiovascular system might contribute to the various aspects of cardiovascular ageing. Therefore, targeting supernumerary senescent cells might be a promising approach to delaying age-dependent decline in cardiovascular health.
总而言之,这些发现表明,心血管系统中不断积累的具有分泌功能和代谢活性的衰老细胞,可能与心血管老化的各个方面有关。因此,靶向过多的衰老细胞可能是一种有前景的方法,可以延缓与年龄相关的心血管健康下降。
Hallmark 7: dysregulated neurohormonal signalling
标志 7:神经激素信号失调
Major components of both systemic and local neurohormonal signalling, including the renin–angiotensin–aldosterone system (RAAS), β-adrenergic signalling and insulin–IGF1 signalling, are chronically activated in ageing, thereby contributing to dysregulation of the cardiovascular system.
包括肾素-血管紧张素-醛固酮系统 (RAAS)、β-肾上腺素能信号传导和胰岛素-IGF1 信号传导在内的全身和局部神经激素信号传导的主要成分,在衰老过程中会被长期激活,从而导致心血管系统失调。
The renin–angiotensin–aldosterone system
肾素-血管紧张素-醛固酮系统
Activation of the RAAS is intimately linked to age-related cardiovascular diseases, including hypertension, atherosclerosis, coronary heart disease, atrial fibrillation and heart failure300. Therefore, RAAS blockade with angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers and mineralocorticoid receptor antagonists is widely used for the treatment of cardiovascular disorders (Table 2). Among patients with obesity and type 2 diabetes mellitus, the beneficial effect of dietary restriction on cardiovascular and renal risk was also linked to suppressed RAAS activity as indicated by reduced serum levels of angiotensin II (the major effector peptide of the classical RAAS)301.
肾素-血管紧张素-醛固酮系统 (RAAS) 的激活与年龄相关的心血管疾病密切相关,包括高血压、动脉粥样硬化、冠心病、房颤和心力衰竭 300 。因此,使用血管紧张素转换酶 (ACE) 抑制剂、血管紧张素受体阻滞剂和盐皮质激素受体拮抗剂阻断 RAAS 广泛用于治疗心血管疾病(表 2)。 在肥胖和 2 型糖尿病患者中,饮食限制对心血管和肾脏的有利影响也与 RAAS 活性受到抑制有关,血清血管紧张素 II(经典 RAAS 的主要效应肽)水平降低就表明了这一点 301 。
Despite its initial adaptive role, chronic activation of the RAAS has adverse cardiovascular effects through various mechanisms, including impaired sodium homeostasis, maladaptive vascular tone and blood volume regulation, coupled with abnormal cellular growth and proliferation, oxidative stress, inflammation and fibrotic remodelling throughout the cardiovascular system302,303. An age-dependent increase in the circulating levels of renin and angiotensin II has been detected in aged but otherwise healthy mice304. Increased expression of angiotensin II mRNA and protein levels is also found locally in the hearts and vasculature of aged mice304,305. Elevated levels of angiotensin II are accompanied by increased vascular expression of ACE, collagen IV, fibronectin and transforming growth factor-β (TGFβ)304 as well as by a greater propensity to develop hypertension in response to low doses of angiotensin II306.
尽管 RAAS 最初具有适应性作用,但慢性激活会通过多种机制对心血管产生不良影响,包括钠稳态受损、血管张力和血容量调节失调,以及整个心血管系统内异常的细胞生长和增殖、氧化应激、炎症和纤维化重塑 302,303 。 在年老的但其他方面健康的 mice 体内,已经检测到循环中肾素和血管紧张素 II 水平随年龄增长而增加 304 。在年老 mice 的心脏和血管中,也发现了血管紧张素 II mRNA 和蛋白水平的局部表达增加 304,305 。 血管紧张素 II 水平升高伴随着血管中 ACE、胶原蛋白 IV、纤连蛋白和转化生长因子-β (TGFβ) 的表达增加 304 ,以及在低剂量血管紧张素 II 刺激下更容易发生高血压 306 。
These studies indicate that RAAS overactivation occurs as a correlate of ageing itself, independently from associated cardiovascular diseases. This notion is further supported by the finding that angiotensin II affects the activity of fundamental pathways involved in organismal ageing (including mTOR, sirtuin, klotho and 5′-AMP-activated protein kinase signalling307), indicating that RAAS activation might be sufficient to promote typical features of cardiovascular ageing. Therefore, in a mouse model, a gain-of-function mutation in the gene encoding the angiotensin II receptor type 1A (AT1A; Agtr1a) causes cardiac fibrosis and diastolic dysfunction, coupled with moderate hypertension308. Even when activated mainly in extrarenal tissues, the RAAS promotes hypertension, cardiac hypertrophy, myocardial fibrosis and diastolic dysfunction in rats at a very young age309. Moreover, in mice, mineralocorticoid receptor blockade improves diastolic dysfunction309, independently of peripheral changes in systolic blood pressure. Further supporting local RAAS effects, overexpression of ACE or angiotensin II in mice, specifically in the heart, triggers atrial dilatation, arrhythmias and sudden cardiac death despite near-normal blood pressure and renal function310. Conversely, pharmacological or genetic inhibition of RAAS signalling attenuates age-related cardiovascular alterations. For example, angiotensin II inhibition and genetic disruption of AT1A delay the onset of cardiovascular decline and improve survival in both mice and rats311,312. Long-term treatment with the ACE inhibitor enalapril attenuates cardiovascular remodelling313, inflammation and frailty in old normotensive mice314. Along similar lines, mice lacking mineralocorticoid receptors in vascular smooth muscle cells avoid the myocardial thickening that usually accompanies ageing and have improved regulation of vascular tone315. Of note, classical RAAS signalling, mediated by angiotensin II, is counteracted by non-canonical RAAS peptides, including angiotensin (1–7), angiotensin (1–9) and alamandine as well as their receptors, which confer protection against cardiovascular disease316. Therefore, despite the widespread clinical use of RAAS inhibitors, the exact contribution of the RAAS to cardiovascular ageing is yet to be fully elucidated.
这些研究表明,RAAS 过度激活是衰老本身的相关因素,与相关的心血管疾病无关。血管紧张素 II 会影响参与机体衰老的基本途径(包括 mTOR、sirtuin、klotho 和 5'-AMP 激活的蛋白激酶信号 307 ),这一发现进一步支持了这一观点,表明 RAAS 激活可能足以促进典型的心血管衰老特征。 因此,在小鼠模型中,编码血管紧张素 II 受体 1A 型(AT1A; Agtr1a)基因的功能获得性突变会导致心脏纤维化和舒张功能障碍,并伴有中度高血压 308 。即使主要在肾外组织中激活,RAAS 也会在非常年轻的大鼠中促进高血压、心脏肥大、心肌纤维化和舒张功能障碍 309 。 此外,在小鼠中,盐皮质激素受体阻断可改善舒张功能障碍 309 ,且与收缩压的外周变化无关。为了进一步支持局部 RAAS 的作用,在小鼠中,特别是在心脏中,ACE 或血管紧张素 II 的过度表达会引发心房扩张、心律失常和心源性猝死,尽管血压和肾功能接近正常 310 。 相反,对 RAAS 信号通路的药物或基因抑制可以减轻与年龄相关的心血管改变。例如,血管紧张素 II 抑制和 AT1A 的基因破坏可以延缓小鼠和大鼠心血管功能下降的发生,并提高存活率 311,312 。用 ACE 抑制剂依那普利进行长期治疗可以减轻老年正常血压小鼠的心血管重构 313 、炎症和体弱 314 。 与之类似,血管平滑肌细胞中缺乏盐皮质激素受体的小鼠可以避免通常伴随衰老发生的心肌增厚,并且血管张力的调节能力有所提高 315 。值得注意的是,由血管紧张素 II 介导的经典 RAAS 信号传导会被非经典 RAAS 肽(包括血管紧张素 (1-7)、血管紧张素 (1-9) 和丙氨酸以及它们的受体)所抵消,这些物质可以预防心血管疾病 316 。 因此,尽管肾素-血管紧张素-醛固酮系统 (RAAS) 抑制剂已广泛应用于临床,但 RAAS 对心血管衰老的具体影响尚待充分阐明。
β-Adrenergic signalling β-肾上腺素能信号传导
Ageing is associated with increased circulating levels of catecholamines, leading to the chronic stimulation and ensuing inactivation of cardiac β-adrenergic receptors, which is aggravated by the age-dependent attrition of cardiac neurons317,318. Indeed, both older humans and rodents have impaired cardiac autonomic regulation, known as β-adrenergic desensitization319, coupled with reduced cardiac reserve and effort intolerance320,321. Similarly, high circulating levels of catecholamines, reduced β-adrenergic responsiveness and general hyposensitivity to adrenergic stress are typically found in human heart failure319. In this context, the suppression of β-adrenergic signalling might be viewed as a compensatory mechanism through which the heart avoids exaggerated responses to high levels of catecholamines. Supporting this notion, activating β-adrenergic signalling in mice by overexpressing β-adrenergic receptors initially improves contractility322,323 but eventually accelerates cardiac hypertrophy and fibrosis, causing fatal cardiac decompensation322,324,325,326. By contrast, several clinical studies have demonstrated that β-blockers improve outcomes in patients with heart failure and reduced ejection fraction327,328,329 (Table 2), and they also attenuate the progression of atherosclerosis330.
衰老与循环儿茶酚胺水平的升高有关,导致心脏β-肾上腺素受体受到慢性刺激并随之失活,而心脏神经元的年龄依赖性损耗会加剧这种情况 317,318 。事实上,老年人和老年啮齿动物都存在心脏自主神经调节障碍,即β-肾上腺素受体脱敏 319 ,同时伴有心脏储备减少和运动不耐受 320,321 。 类似地,在人类心力衰竭中通常可以发现高循环水平的儿茶酚胺、降低的β-肾上腺素能反应性和对肾上腺素能应激的一般低敏感性 319 。在这种情况下,抑制β-肾上腺素能信号传导可能被视为一种补偿机制,通过该机制,心脏可以避免对高水平儿茶酚胺产生过度反应。 为了支持这一观点,通过过度表达β-肾上腺素能受体激活小鼠的β-肾上腺素能信号传导最初会改善收缩力 322,323 ,但最终会加速心脏肥大和纤维化,导致致命的心脏失代偿 322,324,325,326 。相比之下,多项临床研究表明,β受体阻滞剂可以改善射血分数降低的心力衰竭患者的预后 327,328,329 (表 2),并且还可以减缓动脉粥样硬化的进展 330 。
Intriguingly, β-blockade improves, rather than aggravates, impaired β-adrenergic signalling, thereby ameliorating cardiac inotropic reserve in aged but otherwise healthy rats331. Treatment with various β-blockers also extends the median lifespan of mice332. Similarly, genetic inactivation of β-adrenergic signalling by knockout of Adcy5, encoding adenylate cyclase type 5 (AC5; a key enzyme that catalyses the synthesis of cAMP from ATP), in mice prevents age-related cardiac hypertrophy, systolic dysfunction, apoptosis and fibrosis, resulting in increased lifespan333. The beneficial effects of Adcy5 knockout were attributed, at least in part, to the upregulation of superoxide dismutase, which protects against oxidative stress by reducing mitochondrial ROS production333. Emerging evidence also indicates that upregulation of myc box-dependent-interacting protein 1 (also known as bridging integrator 1 (BIN1) or amphiphysin II) might contribute to deranged β-adrenergic signalling during ageing through impaired recycling and trafficking of cardiac Ca2+ channels (published in a preprint334). Accordingly, Bin1 knockdown in old mice improves β-adrenergic responsiveness in the heart as assessed by measuring calcium fluxes in isolated cardiomyocytes334. However, whether suppressing BIN1 would restore inotropic reserve in vivo and could, therefore, be targeted to correct the age-related derangement of β-adrenergic signalling in patients who do not tolerate or benefit from β-blockade is unknown335,336. In essence, fine-tuning β-adrenergic signalling remains a primary objective for the management of cardiovascular risk in older individuals.
有趣的是,β受体阻滞剂可以改善,而不是加重受损的β-肾上腺素能信号传导,从而改善老年但其他方面健康的鼠的心脏变力储备 331 。用各种β受体阻滞剂治疗还可以延长小鼠的平均寿命 332 。 同样地,在小鼠中,通过敲除编码腺苷酸环化酶 5 型(AC5;一种催化 cAMP 由 ATP 合成的关键酶)的 Adcy5 来使β-肾上腺素能信号传导发生遗传性失活,可以防止与年龄相关的心脏肥大、收缩功能障碍、细胞凋亡和纤维化,从而延长寿命 333 。 Adcy5 敲除的有益效果至少部分归因于超氧化物歧化酶的上调,后者通过减少线粒体 ROS 的产生来抵御氧化应激 333 。 新出现的证据也表明,myc 盒依赖性相互作用蛋白 1(也称为桥接整合子 1 (BIN1)或两性蛋白 II)的上调可能通过损害心脏 Ca 2+ 通道的循环和运输(发表在预印本 334 中),从而导致衰老过程中β-肾上腺素信号传导紊乱。因此,在老年小鼠中敲低 Bin1 可以改善心脏中β-肾上腺素的反应性,这可以通过测量分离的心肌细胞中的钙流量来评估 334 。 然而,抑制 BIN1 是否能在体内恢复变力储备,并因此可以作为靶点来纠正对β受体阻滞剂不耐受或无益的患者中与年龄相关的β-肾上腺素能信号紊乱,这一点尚不清楚 335,336 。本质上,微调β-肾上腺素能信号仍然是老年人管理心血管风险的首要目标。
Growth signalling 生长信号
Although the levels of growth hormone and IGF1 naturally decline with age337, mutations resulting in reduced IGF1 signalling are linked to extended lifespan across species71,338. In this regard, the effect of IGF1 signalling on cardiac health seems to be biphasic and age dependent19. Young mice overexpressing the IGF1 receptor (IGF1R) specifically in cardiomyocytes have improved cardiac function and structure19,339, whereas old mice overexpressing cardiac IGF1 or IGF1R have accentuated cardiac ageing, premature heart failure and reduced lifespan19,340. Mechanistically, reduced cardiac healthspan in IGF1R-overexpressing mice is attributable to disabled autophagy associated with mitochondrial dysfunction, reduced oxidative metabolism and impaired myocardial bioenergetics19. By contrast, cardiac IGF1R expression was shown to increase spontaneously with ageing19,341, and its genetic ablation in mice reduces age-related cardiac hypertrophy, fibrosis and levels of pro-inflammatory cytokines341. Late-in-life treatment of wild-type mice with IGF1R monoclonal antibodies improves age-dependent cardiac dysfunction and extends lifespan, albeit only in female animals342. That said, inhibiting cardiac IGF1R signalling in male mice expressing a dominant-negative p110α isoform of phosphatidylinositol 3-kinase (PI3K; also known as phosphoinositide 3-kinase) extends maximum lifespan and attenuates age-related cardiac decline19,343, despite delaying cardiac growth in early life19. Similarly, mice treated with alpelisib, a small-molecule inhibitor of PI3K, have extended median and maximal lifespans in male animals and more so in female animals, despite adverse effects on bone density and glycaemic control344. Whether alpelisib also delays cardiovascular ageing remains to be tested as mice in the study were not assessed at an old age (that is, older than 18 months)344.
虽然生长激素和 IGF1 的水平会随着年龄的增长而自然下降 337 ,但导致 IGF1 信号减少的突变与跨物种的寿命延长有关 71,338 。在这方面,IGF1 信号对心脏健康的影响似乎是双相的,并且是年龄依赖性的 19 。 在心肌细胞中特异性过表达 IGF1 受体(IGF1R)的幼鼠具有改善的心脏功能和结构 19,339 ,而过表达心脏 IGF1 或 IGF1R 的老年小鼠则具有加重的心脏衰老、过早的心力衰竭和缩短的寿命 19,340 。从机制上讲,IGF1R 过表达小鼠的心脏健康寿命缩短归因于与线粒体功能障碍、氧化代谢减少和心肌生物能量学受损相关的自噬功能障碍 19 。 相比之下,研究表明,心脏 IGF1R 的表达会随着年龄的增长而自发增加 19,341 ,在小鼠中基因敲除 IGF1R 可以减少与年龄相关的心脏肥大、纤维化和促炎细胞因子的水平 341 。用 IGF1R 单克隆抗体对野生型小鼠进行晚年治疗,可以改善与年龄相关的心脏功能障碍,并延长寿命,尽管仅在雌性动物中有效 342 。 尽管如此,在表达磷酸肌醇 3-激酶 (PI3K;也称为磷脂酰肌醇 3-激酶) 的显性失活 p110α 亚型的雄性小鼠中抑制心脏 IGF1R 信号传导,可以延长最长寿命并减轻与年龄相关的心脏衰退 19,343 ,尽管这会延缓早期生活中的心脏生长 19 。 同样地,用 PI3K 小分子抑制剂 alpelisib 处理的小鼠,尽管对骨密度和血糖控制有不利影响,但雄性动物的中位和最大寿命都延长了,雌性动物的延长幅度更大 344 。 Alpelisib 是否也会延缓心血管衰老仍有待检验,因为研究中的小鼠未在老年(即大于 18 个月)时进行评估 344 。
As in aged mouse hearts19,341, human explanted failing hearts have elevated IGF1R expression19. Furthermore, cardiac IGF1R mRNA is overexpressed in patients with ischaemic heart disease or HFpEF345. However, the idea that IGF1 signalling could be targeted to decelerate human cardiac ageing is complicated by the observation that both low and high circulating levels of IGF1 are linked to an increased risk of heart failure346,347 and death348,349,350. Therefore, further studies in humans are required to explore the relationship between circulating IGF1 levels and activation of IGF1R signalling in the heart and other tissues to design novel interventions targeting the IGF1–IGF1R system in specific organs.
与老年小鼠心脏相似 19,341 ,在人体移植的衰竭心脏中,IGF1R 表达升高 19 。此外,缺血性心脏病或 HFpEF 患者的心脏 IGF1R mRNA 过度表达 345 。然而,针对 IGF1 信号通路以减缓人类心脏衰老的想法,因以下观察结果而变得复杂:低循环水平和高循环水平的 IGF1 都与心力衰竭 346,347 和死亡 348,349,350 风险增加有关。 因此,还需要进一步的人体研究来探索循环 IGF1 水平与心脏和其他组织中 IGF1R 信号激活之间的关系,从而设计出针对特定器官中 IGF1-IGF1R 系统的新型干预措施。
In the vasculature, declining circulating IGF1 levels correlate with reduced cerebral blood flow and neurovascular coupling in healthy older people (mean age 67.9 years)351. Low serum IGF1 levels in middle-aged women (mean age 52 years) with rheumatoid arthritis are associated with hypertension and an increased risk of cardiovascular events352. Furthermore, in Ames dwarf and Little mice, reduced IGF1 levels are associated with increased vascular stiffness and endothelial dysfunction at a young age, despite increased longevity353,354. Although these observations suggest that IGF1 deficiency actually promotes premature ageing of the vasculature, both low and high levels of IGF1 are associated with an elevated risk of vascular events (a composite of myocardial infarction and stroke) in humans349. Furthermore, global and endothelial cell-specific knockdown of Igf1r in mice improves vascular function at least at a young age355,356. Although these salutary effects await confirmation in aged mice, knockdown of endothelial Igf1r exacerbates atherosclerotic lesions in Apoe−/− mice356. Therefore, long-term studies — preferably focusing on IGF1R modulation in specific vascular cell types — are needed to elucidate the potential pathogenic implications of insufficient versus excessive IGF1 signalling. Of note, another trophic factor, namely VEGF, has been shown to be required for healthy vascular ageing20. Transgenic overexpression of VEGF, which avoids the natural age-related decline in VEGF signalling, reduces endothelial cell senescence, inflammation and mitochondrial dysfunction, promotes blood perfusion of various tissues, and extends healthspan and lifespan20.
在血管系统中,循环 IGF1 水平的下降与健康老年人(平均年龄 67.9 岁)脑血流量和神经血管耦合的降低相关 351 。中年女性(平均年龄 52 岁)类风湿性关节炎患者的低血清 IGF1 水平与高血压和心血管事件风险增加有关 352 。 此外,在艾姆斯矮鼠和小鼠中,即使寿命延长 353,354 ,IGF1 水平降低也与年轻时血管僵硬度和内皮功能障碍有关。尽管这些观察结果表明,IGF1 缺乏实际上会促进血管过早老化,但在人类中,低水平和高水平的 IGF1 都与血管事件(心肌梗死和中风的复合事件)风险升高有关 349 。 此外,在小鼠中进行全身性和内皮细胞特异性的 Igf1r 敲低,至少在年轻时可以改善血管功能 355,356 。尽管这些有益效果有待在老年小鼠中得到证实,但内皮 Igf1r 的敲低会加剧 Apoe −/− 小鼠的动脉粥样硬化病变 356 。 因此,需要进行长期研究——最好侧重于特定血管细胞类型中 IGF1R 的调节——以阐明 IGF1 信号不足与过度潜在的致病意义。值得注意的是,另一种营养因子,即 VEGF,已被证明是健康血管衰老所必需的 20 。 VEGF 的转基因过表达可以避免 VEGF 信号传导自然发生的年龄相关性下降,从而减少内皮细胞衰老、炎症和线粒体功能障碍,促进各种组织的血液灌注,并延长健康寿命和寿命 20 。
Hallmark 8: inflammation 标志 8:炎症
Ageing is associated with chronic low-grade sterile inflammation or inflammageing357. This phenomenon is particularly evident in age-related cardiovascular diseases such as atherosclerosis358. Indeed, atherosclerosis is an inflammatory lesion that smoulders over decades and involves a variety of immune cell subtypes and cytokines that, in combination with risk factors such as dyslipidaemia, contribute to vascular remodelling and plaque formation and rupture203,359. Circulating biomarkers of inflammation predict the progression of atherosclerosis with its cardiovascular complications360, and inflammatory processes apparently further drive secondary events indicative of disease progression361,362. Inflammation and altered immune cell phenotypes are also evident in the ageing heart. The abundance, subtype composition, function and activation state of myocardium-infiltrating immune cells change with time. For example, cardiac myeloid cell numbers that increase in aged mice and humans outnumber lymphocytes, especially in females363,364,365. Myocardial T cell subsets also change with ageing, shifting to a more pro-inflammatory phenotype as exemplified by an increase in CD4+FOXP3–IFNγ+ cells366. Adoptive transfer of differentiated human CD4+ T cells into young mice causes cardiac inflammatory shifts that are typically observed at an old age367. Similarly, T cells transferred from old to young mice show cardiotropism and induce mild dysfunction366, whereas adult T cells transferred into neonates promote local inflammation and impair cardiac regenerative capacity368. More dramatically, T cell-specific deficiency in mitochondrial transcription factor A promotes chronic inflammation and accelerates organismal ageing, including in the cardiovascular system, which can be attenuated by TNF neutralization369. Therefore, immunosenescence affecting T lymphocytes can accelerate cardiovascular ageing.
衰老与慢性低度无菌性炎症或炎症衰老相关 357 。这种现象在与年龄相关的心血管疾病(如动脉粥样硬化)中尤为明显 358 。事实上,动脉粥样硬化是一种潜伏数十年的炎症病变,涉及多种免疫细胞亚型和细胞因子,这些因素与血脂异常等危险因素相结合,会导致血管重塑、斑块形成和破裂 203,359 。 循环炎症生物标志物可以预测动脉粥样硬化的进展及其心血管并发症 360 ,而且炎症过程显然会进一步驱动表明疾病进展的二次事件 361,362 。炎症和免疫细胞表型的改变在衰老的心脏中也很明显。随着时间的推移,心肌浸润免疫细胞的丰度、亚型组成、功能和激活状态会发生变化。 例如,在老年小鼠和人类中,心脏髓系细胞的数量会增加,超过淋巴细胞的数量,尤其是在女性中 363,364,365 。心肌 T 细胞亚群也会随着衰老而发生变化,转变为更促炎的表型,例如 CD4 + FOXP3 – IFNγ + 细胞的增加 366 。将分化的人类 CD4 + T 细胞过继转移到幼鼠体内会导致通常在老年时观察到的心脏炎症转移 367 。 类似地,从老年小鼠转移到幼年小鼠的 T 细胞表现出嗜心性并诱导轻度功能障碍 366 ,而转移到新生儿体内的成年 T 细胞会促进局部炎症并损害心脏再生能力 368 。更引人注目的是,T 细胞特异性线粒体转录因子 A 缺陷会促进慢性炎症并加速机体衰老,包括心血管系统,而 TNF 中和可以减缓这一过程 369 。 因此,影响 T 淋巴细胞的免疫衰老会加速心血管衰老。
Cardiac-specific induction of inflammation by a gain-of-function mutation in the gene encoding IκB kinase, which activates NF-κΒ, causes spontaneous cardiac hypertrophy in mice, coupled to early-onset heart failure and premature mortality370. Transgenic mice overexpressing TNF in the heart also have signs of hypertrophic cardiomyopathy at a young age371. Conversely, mice lacking the inflammasome component NLRP3 have an extended lifespan and delayed cardiac ageing, coinciding with reduced IGF1 signalling, increased autophagy, and upregulation of SIRT1 and the NAD+ salvage enzyme nicotinamide phosphoribosyltransferase372. Similarly, transgenic expression of a dominant-negative form of IκB in endothelial cells reduces vascular senescence and extends lifespan373. In patients with cardiovascular disease, targeting inflammation by either inhibiting the NLPR3 inflammasome with colchicine374 or neutralizing its major cytokine product IL-1β with the anti-IL-1β antibody canakinumab375,376, has been shown to be effective for secondary prevention of cardiovascular events. Lung cancer mortality was also reduced in patients with atherosclerosis receiving canakinumab377, although at the cost of an increased incidence of sometimes fatal infections375.
编码 IκB 激酶(可激活 NF-κΒ)的基因发生功能获得性突变,从而导致心脏特异性炎症的诱导,会导致小鼠出现自发性心脏肥大,并伴有早发性心力衰竭和过早死亡 370 。在心脏中过度表达 TNF 的转基因小鼠在年轻时也表现出肥厚型心肌病 371 的迹象。 相反,缺乏炎症小体成分 NLRP3 的小鼠寿命延长,心脏衰老延迟,同时伴随着 IGF1 信号的减少、自噬的增加以及 SIRT1 和 NAD + 补救酶烟酰胺磷酸核糖转移酶 372 的上调。类似地,内皮细胞中显性失活型 IκB 的转基因表达可以减少血管衰老并延长寿命 373 。 在心血管疾病患者中,通过用秋水仙碱 374 抑制 NLPR3 炎症小体或用抗 IL-1β抗体卡纳单抗 375,376 中和其主要细胞因子产物 IL-1β来靶向炎症,已被证明对心血管事件的二级预防有效。接受卡纳单抗治疗的动脉粥样硬化患者的肺癌死亡率也降低了 377 ,但代价是感染的发生率增加,有时甚至是致命的 375 。
In addition to atherosclerosis and related coronary artery disease, HFpEF (the predominant form of cardiac failure in older individuals378) has also been proposed to be an inflammatory, multiorgan syndrome379, primarily driven by accelerated ageing380,381. Supporting this notion, older individuals (mean age 73.6 years) with elevated levels of biomarkers of inflammation have a significantly higher risk of HFpEF than those with normal levels of inflammatory biomarkers382. Of note, the cardioprotective effects of sodium–glucose cotransporter 2 (SGLT2) inhibitors — the first drug class showing efficacy in the treatment of HFpEF383,384 — are linked to reduced inflammation385,386,387, among other anti-ageing effects387,388,389. In aged mice fed a high-fat diet and desoxycorticosterone, the development of HFpEF can be prevented by the administration of β-hydroxybutyrate, which acts directly on the NLPR3 inflammasome to inhibit IL-1β maturation and pro-inflammatory, cytokine-induced mitochondrial dysfunction390.
除了动脉粥样硬化和相关冠状动脉疾病外,HFpEF(老年人中最常见的心力衰竭形式 378 )也被认为是一种炎症性多器官综合征 379 ,主要由加速衰老驱动 380,381 。支持这一观点的是,炎症生物标志物水平升高的老年人(平均年龄 73.6 岁)患 HFpEF 的风险显著高于炎症生物标志物水平正常的人 382 。 值得注意的是,钠-葡萄糖协同转运蛋白 2 (SGLT2) 抑制剂——第一个在 HFpEF 治疗中显示出疗效的药物 383,384 ——的心脏保护作用与其他抗衰老作用 387,388,389 一样,与炎症的减少有关 385,386,387 。 在高脂饮食和去氧皮质酮喂养的衰老小鼠中,HFpEF 的发生可以通过施用β-羟基丁酸来预防,β-羟基丁酸直接作用于 NLPR3 炎症小体,抑制 IL-1β的成熟和促炎、细胞因子诱导的线粒体功能障碍 390 。
Mechanistically, cardiovascular ‘inflammageing’ is intrinsically linked with other hallmarks of ageing, including disabled macroautophagy (via accumulation of harmful waste), loss of proteostasis (via a disequilibrium between pro-inflammatory and anti-inflammatory proteins), genomic instability (via CHIP), epigenetic alterations (via promotion of pro-inflammatory gene transcription), mitochondrial dysfunction (via excessive ROS) and cell senescence (via SASP). Notably, the innate cGAS–STING immune pathway, which senses cytosolic DNA and promotes downstream activation of interferon-dependent inflammatory pathways, is crucial for cellular senescence and SASP acquisition391. Therefore, the age-dependent decline in transcriptional coactivator YAP–TAZ mechanosignalling, evident in fibroblasts, vascular smooth muscle cells and cardiomyocytes, promotes senescence in the mouse aorta via cGAS–STING induction392. cGAS–STING signalling is also increased in association with age-related endothelial dysfunction in aged mice and humans231 as well as in the premature ageing seen with progeria (also known as Hutchinson–Gilford syndrome)393, in which cardiovascular ageing manifests during childhood or early adolescence394. Notably, in mice, genetic or pharmacological inhibition of cGAS–STING ameliorates the phenotypes of atherosclerosis, myocardial infarction and stroke395,396,397,398,399.
从机制上讲,心血管“炎症衰老”与其他衰老特征内在相关,包括巨自噬功能障碍(通过有害废物的积累)、蛋白稳态丧失(通过促炎和抗炎蛋白之间的失衡)、基因组不稳定性(通过 CHIP)、表观遗传改变(通过促进促炎基因转录)、线粒体功能障碍(通过过多的 ROS)和细胞衰老(通过 SASP)。 值得注意的是,先天性 cGAS–STING 免疫通路对胞质 DNA 进行感应,并促进下游干扰素依赖性炎症通路的激活,这对于细胞衰老和 SASP 的获得至关重要 391 。因此,在成纤维细胞、血管平滑肌细胞和心肌细胞中显而易见的转录共激活因子 YAP–TAZ 机械信号传导的年龄依赖性下降,通过 cGAS–STING 的诱导促进小鼠主动脉的衰老 392 。 cGAS–STING 信号通路也与老年小鼠和人类的年龄相关性内皮功能障碍有关 231 ,并且与早衰症(也称为 Hutchinson–Gilford 综合征)的早衰现象有关 393 ,在早衰症中,心血管老化在儿童期或青春期早期表现出来 394 。值得注意的是,在小鼠中,cGAS–STING 的基因或药物抑制可以改善动脉粥样硬化、心肌梗死和中风的表型 395,396,397,398,399 。
Although innate immunity was traditionally considered to be the main driver of inflammation in cardiovascular ageing and disease, adaptive immunity has also been implicated in these processes. With ageing, immunosenescence, T cell exhaustion and other signs of adaptive immune dysfunction not only explain insufficient immune responses to external or internal pathogens but also contribute to systemically increased inflammation, which drives infections, cancer and cardiovascular disease400.
尽管传统上认为先天免疫是心血管衰老和疾病炎症的主要驱动因素,但适应性免疫也与这些过程有关。随着衰老、免疫衰老、T 细胞耗竭和其他适应性免疫功能障碍的迹象不仅解释了对外部或内部病原体的免疫反应不足,而且还有助于全身性炎症的增加,从而驱动感染、癌症和心血管疾病 400 。
Despite the broad evidence base implicating inflammation and immune mechanisms in the pathogenesis of age-associated cardiovascular diseases in preclinical models, anti-inflammatory drugs have not yet been convincingly shown to mediate the primary prevention of cardiovascular events in humans22. For example, low-dose aspirin, a non-steroidal anti-inflammatory drug that also inhibits platelet aggregation and induces autophagy401, has not been shown to prevent initial cardiovascular events in large clinical trials402,403,404. Whether this failure is due to low adherence or the need to combine aspirin with other drugs is a matter of debate. Indeed, in 2022, a polypill including aspirin was shown to improve both adherence and efficacy in secondary prevention of cardiovascular disease405, but these effects remain to be examined in the setting of primary prevention. In addition, whether all individuals beyond a certain age should be treated with anti-inflammatory drugs to prevent cardiovascular ageing or whether only those with subclinical features of inflammation should be targeted is uncertain. For example, older individuals harbouring CHIP mutations might be particularly responsive to IL-1β neutralization by canakinumab as suggested by a post hoc analysis of the CANTOS trial406. Canakinumab therapy was particularly effective in patients with signs of systemic inflammation (plasma levels of high-sensitivity CRP ≥2 mg/l), leading to an early and robust reduction in inflammatory markers407.
尽管有广泛的证据表明炎症和免疫机制与临床前模型中年龄相关的心血管疾病的发病机制有关,但尚未令人信服地证明抗炎药物可以介导人类心血管事件的一级预防 22 。 例如,低剂量阿司匹林是一种非甾体抗炎药,也可抑制血小板聚集并诱导自噬 401 ,但在大型临床试验中,尚未证实其能预防初始心血管事件 402,403,404 。这种失败是由于患者依从性低,还是需要将阿司匹林与其他药物联合使用,这是一个有争议的问题。 事实上,在 2022 年,一项包含阿司匹林的复方制剂被证实可以提高心血管疾病二级预防的依从性和有效性 405 ,但这些效果仍有待在一级预防的背景下进行检验。此外,是否所有超过特定年龄的人都应该接受抗炎药物治疗以预防心血管衰老,或者是否只应针对那些具有亚临床炎症特征的人,目前尚不确定。 例如,携带 CHIP 突变的老年人可能对卡纳单抗中和 IL-1β特别敏感,正如 CANTOS 试验的事后分析所表明的 406 。卡纳单抗疗法在有全身炎症迹象(血浆中高敏 CRP 水平≥2 mg/l)的患者中尤其有效,导致炎症标志物的早期和显著减少 407 。
As discussed, abundant evidence exists to support the idea that inflammatory processes contribute to cardiac and vascular ageing. However, which patient populations will benefit from anti-inflammatory interventions, and whether simultaneous targeting of several cell types (such as macrophages and T lymphocytes), intracellular pathways (such as the cGAS–STING, NF-κB and inflammasome pathways) or pro-inflammatory effector molecules (such as type I interferons, TNF and IL-1β) might constitute an effective combination strategy for postponing or halting cardiovascular ageing remains to be determined.
如前所述,大量证据表明炎症过程促进了心脏和血管的衰老。 然而,哪些患者群体将受益于抗炎干预,以及同时靶向多种细胞类型(如巨噬细胞和 T 淋巴细胞)、细胞内通路(如 cGAS–STING、NF-κB 和炎性体通路)或促炎效应分子(如 I 型干扰素、TNF 和 IL-1β)是否可能构成一种有效的联合策略,以延缓或阻止心血管衰老,仍有待确定。
Conclusions 结论
The hallmarks of cardiovascular ageing are strongly intertwined, meaning that the experimental accentuation or attenuation of any individual hallmark affects most, if not all, of the others. For instance, disrupted autophagy leads to proteostatic collapse, mitochondrial dysfunction, dysregulated neurohormonal signalling and increased inflammation26,29. Conversely, experimental induction of autophagy also improves mitochondrial function, protects against proteostasis loss, reduces cell senescence and attenuates inflammation237,408,409. Similarly, genomic instability is a major driver of cell senescence, which subsequently promotes inflammation through SASP. Genetic instability also directly promotes inflammation through CHIP410. Another example is provided by epigenetic alterations in ncRNA and histones that disable autophagy, induce cell senescence and disrupt various neurohormonal signalling pathways in the ageing cardiovascular system.
心血管衰老的标志紧密相连,这意味着对任何单个标志的实验性强化或减弱都会影响到大多数(如果不是全部)其他标志。例如,自噬中断会导致蛋白稳态崩溃、线粒体功能障碍、神经内分泌信号失调和炎症增加 26,29 。 相反,实验诱导的自噬也能改善线粒体功能,防止蛋白稳态丧失,减少细胞衰老并减弱炎症 237,408,409 。同样,基因组不稳定性是细胞衰老的主要驱动因素,随后通过 SASP 促进炎症。基因不稳定性也通过 CHIP 直接促进炎症 410 。 非编码 RNA 和组蛋白的表观遗传改变是另一个例子,它们会使自噬失效、诱导细胞衰老并破坏衰老心血管系统中的各种神经激素信号通路。
We propose a didactic distinction between the hallmarks of cardiovascular ageing whereby the first four hallmarks discussed in this Review (disabled macroautophagy, loss of proteostasis, genomic instability and epigenetic alterations), which progressively advance with age and underlie the deterioration of the genome, epigenome, proteome and organelles, should be considered as primary hallmarks that initiate cardiovascular ageing at the upstream level. Further downstream, we posit that mitochondrial dysfunction, dysregulated neurohormonal signalling and cell senescence are antagonistic hallmarks that reflect (mal)adaptive responses to damage and therefore have a more nuanced role in cardiovascular ageing. Finally, when the accumulated damage inflicted by the primary and antagonistic hallmarks reaches a threshold level, chronic low-grade inflammation ensues as an integrative feature of cardiovascular ageing that ultimately precipitates the manifestation of disease (Fig. 2 and Box 3). Notably, persistent exposure to external factors, such as poor nutrition, sedentary lifestyle, psychological and social stress, smoking, and pollution, might also promote the development of cardiovascular disease by acting on these hallmarks at different levels267. This notion is exemplified by the major cardiovascular risk factor of obesity411, in which disabled autophagy412, loss of proteostasis413, epigenetic alterations414, mitochondrial dysfunction415, cell senescence416, neurohormonal signalling dysregulation417,418 and chronic inflammation419 have been reported, and coincide with an accelerated cardiovascular ageing phenotype420,421. By contrast, moderate body weight loss in response to dietary restriction has shown promising cardiovascular and other anti-ageing benefits422, including in mice and humans without obesity423,424,425.
我们提出了一个关于心血管衰老标志的教学区分,其中本综述中讨论的前四个标志(巨自噬功能障碍、蛋白稳态丧失、基因组不稳定性和表观遗传改变)随着年龄的增长而逐渐发展,并导致基因组、表观基因组、蛋白质组和细胞器的恶化,应被视为在上游水平启动心血管衰老的主要标志。 更进一步,我们认为线粒体功能障碍、神经激素信号失调和细胞衰老是对损伤产生(不)良性适应性反应的对抗性标志,因此在心血管衰老中扮演着更为微妙的角色。 最后,当主要和拮抗性标志造成的累积损伤达到阈值水平时,慢性低度炎症就会随之而来,作为心血管衰老的一个综合特征,最终促使疾病的发生(图 2 和框 3)。 值得注意的是,长期暴露于外部因素,如不良营养、久坐的生活方式、心理和社会压力、吸烟和污染,也可能通过在不同层面上作用于这些特征来促进心血管疾病的发生 267 。 肥胖是主要的心血管危险因素 411 ,这体现了上述观点,其中已报道了自噬功能障碍 412 、蛋白稳态丧失 413 、表观遗传改变 414 、线粒体功能障碍 415 、细胞衰老 416 、神经激素信号失调 417,418 和慢性炎症 419 ,并且这些都与加速的心血管衰老表型相吻合 420,421 。 相比之下,通过饮食限制实现适度的体重减轻已显示出令人鼓舞的心血管和其他抗衰老益处 422 ,包括在没有肥胖的小鼠和人类中 423,424,425 。
Despite the efficient management of traditional risk factors, such as dyslipidaemia and hypertension, residual cardiovascular risk remains, suggesting that the conventional management of cardiovascular risk factors has reached a glass ceiling. Indeed, currently available lipid-lowering therapies, including proprotein convertase subtilisin–kexin type 9 (PCSK9) inhibitors, almost eliminate LDL cholesterol from the plasma but do not eradicate cardiovascular risk426, denoting the need for novel strategies targeting one or more of the hallmarks of ageing. Indeed, each of the cardiovascular ageing hallmarks can be considered as a potential target for the development of novel medicines to extend cardiovascular healthspan. Theoretically, the level of interconnectivity of the hallmarks will determine whether drugs targeting distinct features of cardiovascular ageing can be advantageously combined to yield additive, and perhaps even synergistic, effects. Therefore, systematic evaluation of simultaneous interventions on distinct facets of the ageing process will be necessary as will the measurement of outcomes with respect to both cardiovascular and general health. Despite extensive preclinical evidence, additional studies are also needed to consolidate the translational relevance and targetability of the hallmarks of ageing in humans. Although animal models phenocopy many aspects of human ageing, they cannot entirely encompass its complexity. This limitation is particularly relevant for age-related atherosclerotic disease, which is almost inevitable in humans, but must be artificially induced in rodents via specific mutations (such as deletion of Apoe or Ldlr) and feeding with atherogenic diets rich in cholesterol.
尽管对血脂异常和高血压等传统风险因素进行了有效管理,但残余心血管风险仍然存在,这表明对心血管风险因素的常规管理已经达到了瓶颈。 事实上,目前可用的降脂疗法,包括前蛋白转化酶枯草杆菌蛋白酶/kexin 9 型(PCSK9)抑制剂,几乎可以消除血浆中的低密度脂蛋白胆固醇,但并不能根除心血管风险 426 ,这表明需要针对一个或多个衰老特征的新策略。事实上,每个心血管衰老特征都可以被认为是开发延长心血管健康寿命的新药的潜在靶点。 从理论上讲,这些衰老特征的相互关联程度将决定靶向不同心血管衰老特征的药物是否可以有利地结合使用,从而产生累加甚至协同效应。因此,有必要对同时干预衰老过程的不同方面进行系统评估,并衡量其对心血管和整体健康的影响。 尽管有大量的临床前证据,但还需要更多的研究来巩固衰老特征在人类中的转化相关性和可靶向性。虽然动物模型可以在表型上模拟人类衰老的许多方面,但它们无法完全涵盖其复杂性。 这种局限性与年龄相关动脉粥样硬化疾病尤其相关,这种疾病在人类中几乎不可避免,但在啮齿动物中必须通过特定的突变(例如 Apoe 或 Ldlr 的缺失)和富含胆固醇的致动脉粥样硬化饮食来人为诱导。
Beyond pharmacological treatments, an in-depth understanding of the extent to which lifestyle interventions (such as diet and exercise) can be optimized to enable the cardiovascular system to adapt to various environmental stressors and decelerate the ageing process is needed. Indeed, the literature on the molecular basis of healthy diets, regular physical activity and adequate psychosocial stress responses continues to expand108. Therefore, with future research, we should aim to establish objective, biomarker-guided recommendations for the adoption of healthy habits in combination with specific pharmacopreventive strategies.
除了药物治疗之外,还需要深入了解如何优化生活方式干预(如饮食和运动),使心血管系统能够适应各种环境压力,并减缓衰老过程。事实上,关于健康饮食、规律体育活动和充分的社会心理压力反应的分子基础的文献正在不断扩展 108 。 因此,通过未来的研究,我们应该致力于制定客观的、以生物标志物为指导的建议,以便采用健康习惯,并结合特定的药物预防策略。
References 参考文献
Bergmann, O. et al. Evidence for cardiomyocyte renewal in humans. Science 324, 98–102 (2009).
Bergmann, O. 等. 人类心肌细胞更新的证据。科学 324, 98–102 (2009)。Senyo, S. E. et al. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493, 433–436 (2013).
Senyo, S. E. 等. 通过预先存在的心肌细胞实现哺乳动物心脏的更新。自然 493, 433–436 (2013)。Bergmann, O. et al. Dynamics of cell generation and turnover in the human heart. Cell 161, 1566–1575 (2015).
Bergmann, O. 等. 人类心脏中细胞生成和更新的动态。细胞 161, 1566–1575 (2015)。Ji, H. et al. Sex differences in myocardial and vascular aging. Circ. Res. 130, 566–577 (2022).
Ji, H. 等。心肌和血管衰老的性别差异。《循环研究》130, 566–577 (2022)。Ungvari, Z. et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat. Rev. Cardiol. 15, 555–565 (2018).
Ungvari, Z. 等。衰老血管中的内皮功能障碍和血管生成障碍。《自然综述:心脏病学》15, 555–565 (2018)。Ungvari, Z., Tarantini, S., Sorond, F., Merkely, B. & Csiszar, A. Mechanisms of vascular aging, a geroscience perspective: JACC Focus Seminar. J. Am. Coll. Cardiol. 75, 931–941 (2020).
Ungvari, Z.,Tarantini, S.,Sorond, F.,Merkely, B. & Csiszar, A. 血管衰老的机制,一个老年科学的视角:《美国心脏病学会杂志》焦点研讨会. J. Am. Coll. Cardiol. 75, 931–941 (2020).Lakatta, E. G. & Levy, D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a ‘set up’ for vascular disease. Circulation 107, 139–146 (2003).
Lakatta, E. G. & Levy, D. 动脉和心脏衰老:心血管疾病企业的主要股东:第一部分:衰老的动脉:血管疾病的“铺垫”. Circulation 107, 139–146 (2003).Lakatta, E. G. & Levy, D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation 107, 346–354 (2003).
Lakatta, E. G. & Levy, D. 动脉和心脏衰老:心血管疾病企业的主要股东:第二部分:健康衰老的心脏:与心脏病的联系。 Circulation 107, 346–354 (2003).North, B. J. & Sinclair, D. A. The intersection between aging and cardiovascular disease. Circ. Res. 110, 1097–1108 (2012).
North, B. J. & Sinclair, D. A. 衰老与心血管疾病的交集。 Circ. Res. 110, 1097–1108 (2012).Michos, E. D., McEvoy, J. W. & Blumenthal, R. S. Lipid management for the prevention of atherosclerotic cardiovascular disease. N. Engl. J. Med. 381, 1557–1567 (2019).
Michos, E. D., McEvoy, J. W. & Blumenthal, R. S. 脂类管理在动脉粥样硬化性心血管疾病预防中的作用。N. Engl. J. Med. 381, 1557–1567 (2019).Fuchs, F. D. & Whelton, P. K. High blood pressure and cardiovascular disease. Hypertension 75, 285–292 (2020).
Fuchs, F. D. & Whelton, P. K. 高血压与心血管疾病。Hypertension 75, 285–292 (2020).GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 (2020).
GBD 2019 疾病和损伤合作者。204 个国家和地区 369 种疾病和损伤的全球负担,1990-2019 年:2019 年全球疾病负担研究的系统分析。《柳叶刀》396, 1204–1222 (2020)。Lieb, W., Enserro, D. M., Larson, M. G. & Vasan, R. S. Residual cardiovascular risk in individuals on lipid-lowering treatment: quantifying absolute and relative risk in the community. Open Heart 5, e000722 (2018).
Lieb, W.,Enserro, D. M.,Larson, M. G. & Vasan, R. S. 接受降脂治疗的个体中的残余心血管风险:量化社区中的绝对和相对风险。《开放心脏》5, e000722 (2018)。Lieb, W., Enserro, D. M., Sullivan, L. M. & Vasan, R. S. Residual cardiovascular risk in individuals on blood pressure-lowering treatment. J. Am. Heart Assoc. 4, e002155 (2015).
Lieb, W., Enserro, D. M., Sullivan, L. M. & Vasan, R. S. 接受降血压治疗的个体中的残余心血管风险。J. Am. Heart Assoc. 4, e002155 (2015).Vanuzzo, D. The epidemiological concept of residual risk. Intern. Emerg. Med. 6, 45–51 (2011).
Vanuzzo, D. 残余风险的流行病学概念。Intern. Emerg. Med. 6, 45–51 (2011).Moturi, S., Ghosh-Choudhary, S. K. & Finkel, T. Cardiovascular disease and the biology of aging. J. Mol. Cell Cardiol. 167, 109–117 (2022).
Moturi, S., Ghosh-Choudhary, S. K. & Finkel, T. 心血管疾病与衰老生物学。J. Mol. Cell Cardiol. 167, 109–117 (2022).Zhernakova, D. V. et al. Age-dependent sex differences in cardiometabolic risk factors. Nat. Cardiovasc. Res. 1, 844–854 (2022).
Zhernakova, D. V. et al. 心脏代谢风险因素中与年龄相关的性别差异。Nat. Cardiovasc. Res. 1, 844–854 (2022).Hägg, S. & Jylhävä, J. Sex differences in biological aging with a focus on human studies. eLife 10, e63425 (2021).
Abdellatif, M. et al. Fine-tuning cardiac insulin-like growth factor 1 receptor signaling to promote health and longevity. Circulation 145, 1853–1866 (2022).
Grunewald, M. et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science 373, eabc8479 (2021).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Levine, B. & Kroemer, G. Biological functions of autophagy genes: a disease perspective. Cell 176, 11–42 (2019).
Abdellatif, M. & Kroemer, G. Co-ordinated mitochondrial degradation by autophagy and heterophagy in cardiac homeostasis. Cardiovasc. Res. 117, e1–e3 (2021).
Nicolás-Ávila, J. A. et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183, 94–109.e23 (2020).
Abdellatif, M., Sedej, S., Carmona-Gutierrez, D., Madeo, F. & Kroemer, G. Autophagy in cardiovascular aging. Circ. Res. 123, 803–824 (2018).
Abdellatif, M., Ljubojevic-Holzer, S., Madeo, F. & Sedej, S. Autophagy in cardiovascular health and disease. Prog. Mol. Biol. Transl. Sci. 172, 87–106 (2020).
Picca, A. et al. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 15, 543–554 (2018).
Bravo-San Pedro, J. M., Kroemer, G. & Galluzzi, L. Autophagy and mitophagy in cardiovascular disease. Circ. Res. 120, 1812–1824 (2017).
Kanamori, H. et al. Impact of autophagy on prognosis of patients with dilated cardiomyopathy. J. Am. Coll. Cardiol. 79, 789–801 (2022).
Jefferies, J. L. & Saffitz, J. E. Autophagy and reverse remodeling: a new biomarker in heart failure? J. Am. Coll. Cardiol. 79, 802–804 (2022).
Pietrocola, F. et al. Aspirin recapitulates features of caloric restriction. Cell Rep. 22, 2395–2407 (2018).
Farah, B. L. et al. β-Adrenergic agonist and antagonist regulation of autophagy in HepG2 cells, primary mouse hepatocytes, and mouse liver. PLoS One 9, e98155 (2014).
Kania, E. et al. Verapamil treatment induces cytoprotective autophagy by modulating cellular metabolism. FEBS J. 284, 1370–1387 (2017).
Rubinsztein, D. C., Mariño, G. & Kroemer, G. Autophagy and aging. Cell 146, 682–695 (2011).
Kaushik, S. et al. Autophagy and the hallmarks of aging. Ageing Res. Rev. 72, 101468 (2021).
Klionsky, D. J. et al. Autophagy in major human diseases. EMBO J. 40, e108863 (2021).
LaRocca, T. J. et al. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 590, 3305–3316 (2012).
Linton, P.-J., Gurney, M., Sengstock, D., Mentzer, R. M. & Gottlieb, R. A. This old heart: cardiac aging and autophagy. J. Mol. Cell Cardiol. 83, 44–54 (2015).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17, 1–382 (2021).
Cho, J. M. et al. Late-in-life treadmill training rejuvenates autophagy, protein aggregate clearance, and function in mouse hearts. Aging Cell 20, e13467 (2021).
Wu, B. et al. Aldehyde dehydrogenase 2 activation in aged heart improves the autophagy by reducing the carbonyl modification on SIRT1. Oncotarget 7, 2175–2188 (2016).
Chang, K. et al. TGFB-INHB/activin signaling regulates age-dependent autophagy and cardiac health through inhibition of MTORC2. Autophagy 16, 1807–1822 (2020).
Ma, L. et al. Restoring pharmacologic preconditioning in the aging heart: role of mitophagy/autophagy. J. Gerontol. A Biol. Sci. Med. Sci. 72, 489–498 (2017).
Pietrocola, F., Galluzzi, L., Bravo-San Pedro, J. M., Madeo, F. & Kroemer, G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 21, 805–821 (2015).
Kane, A. E. & Sinclair, D. A. Sirtuins and NAD+ in the development and treatment of metabolic and cardiovascular diseases. Circ. Res. 123, 868–885 (2018).
Harraz, O. F. & Jensen, L. J. Vascular calcium signalling and ageing. J. Physiol. 599, 5361–5377 (2021).
Shaikh, S. et al. Regulation of cardiomyocyte autophagy by calcium. Am. J. Physiol. Endocrinol. Metab. 310, E587–E596 (2016).
Behringer, E. J. & Segal, S. S. Impact of aging on calcium signaling and membrane potential in endothelium of resistance arteries: a role for mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 72, 1627–1637 (2017).
Wang, J. et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging 12, 650–671 (2020).
Pucciarelli, S. et al. Spermidine and spermine are enriched in whole blood of nona/centenarians. Rejuvenation Res. 15, 590–595 (2012).
Madeo, F., Eisenberg, T., Pietrocola, F. & Kroemer, G. Spermidine in health and disease. Science 359, eaan2788 (2018).
LaRocca, T. J., Gioscia-Ryan, R. A., Hearon, C. M. & Seals, D. R. The autophagy enhancer spermidine reverses arterial aging. Mech. Ageing Dev. 134, 314–320 (2013).
Eisenberg, T. et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 22, 1428–1438 (2016).
Taneike, M. et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6, 600–606 (2010).
Ljubojević-Holzer, S. et al. Loss of autophagy protein ATG5 impairs cardiac capacity in mice and humans through diminishing mitochondrial abundance and disrupting Ca2+ cycling. Cardiovasc. Res. 118, 1492–1505 (2022).
Michiels, C. F., Fransen, P., De Munck, D. G., De Meyer, G. R. Y. & Martinet, W. Defective autophagy in vascular smooth muscle cells alters contractility and Ca2+ homeostasis in mice. Am. J. Physiol. Heart Circ. Physiol. 308, H557–H567 (2015).
Grootaert, M. O. et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 11, 2014–2032 (2015).
Torisu, T. et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat. Med. 19, 1281–1287 (2013).
Hsieh, P. N. et al. A conserved KLF-autophagy pathway modulates nematode lifespan and mammalian age-associated vascular dysfunction. Nat. Commun. 8, 914 (2017).
Fernández, Á. F. et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 558, 136–140 (2018).
Alfaras, I. et al. Pharmacological strategies to retard cardiovascular aging. Circ. Res. 118, 1626–1642 (2016).
Voglhuber, J., Ljubojevic-Holzer, S., Abdellatif, M. & Sedej, S. Targeting cardiovascular risk factors through dietary adaptations and caloric restriction mimetics. Front. Nutr. 8, 758058 (2021).
Weiss, E. P. & Fontana, L. Caloric restriction: powerful protection for the aging heart and vasculature. Am. J. Physiol. Heart Circ. Physiol. 301, H1205–H1219 (2011).
Madeo, F., Carmona-Gutierrez, D., Hofer, S. J. & Kroemer, G. Caloric restriction mimetics against age-associated disease: targets, mechanisms, and therapeutic potential. Cell Metab. 29, 592–610 (2019).
Quarles, E. et al. Rapamycin persistently improves cardiac function in aged, male and female mice, even following cessation of treatment. Aging Cell 19, e13086 (2020).
Lesniewski, L. A. et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 16, 17–26 (2017).
Kaplon, R. E. et al. Oral trehalose supplementation improves resistance artery endothelial function in healthy middle-aged and older adults. Aging 8, 1167–1183 (2016).
Flynn, J. M. et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell 12, 851–862 (2013).
López-Otín, C. & Kroemer, G. Hallmarks of health. Cell 184, 1929–1939 (2021).
Hipp, M. S., Kasturi, P. & Hartl, F. U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435 (2019).
Kaushik, S. & Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381 (2018).
Alberti, S. & Hyman, A. A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 22, 196–213 (2021).
Gouveia, M., Xia, K., Colón, W., Vieira, S. I. & Ribeiro, F. Protein aggregation, cardiovascular diseases, and exercise training: where do we stand? Ageing Res. Rev. 40, 1–10 (2017).
Henning, R. H. & Brundel, B. J. J. M. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 14, 637–653 (2017).
González-López, E. et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur. Heart J. 36, 2585–2594 (2015).
Rainer, P. P. et al. Desmin phosphorylation triggers preamyloid oligomers formation and myocyte dysfunction in acquired heart failure. Circ. Res. 122, e75–e83 (2018).
Hofmann, C., Katus, H. A. & Doroudgar, S. Protein misfolding in cardiac disease. Circulation 139, 2085–2088 (2019).
McLendon, P. M. & Robbins, J. Proteotoxicity and cardiac dysfunction. Circ. Res. 116, 1863–1882 (2015).
Martin, T. G. et al. Cardiomyocyte contractile impairment in heart failure results from reduced BAG3-mediated sarcomeric protein turnover. Nat. Commun. 12, 2942 (2021).
Bulteau, A.-L., Szweda, L. I. & Friguet, B. Age-dependent declines in proteasome activity in the heart. Arch. Biochem. Biophys. 397, 298–304 (2002).
Colotti, C. et al. Effects of aging and anti-aging caloric restrictions on carbonyl and heat shock protein levels and expression. Biogerontology 6, 397–406 (2005).
Dai, D. et al. Altered proteome turnover and remodeling by short‐term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13, 529–539 (2014).
Ayyadevara, S. et al. Age- and hypertension-associated protein aggregates in mouse heart have similar proteomic profiles. Hypertension 67, 1006–1013 (2016).
Madrigal-Matute, J. et al. Protective role of chaperone-mediated autophagy against atherosclerosis. Proc. Natl Acad. Sci. USA 119, e2121133119 (2022).
Tabula Sapiens Consortium et al. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science 376, eabl4896 (2022).
Marfella, R. et al. Effects of ubiquitin-proteasome system deregulation on the vascular senescence and atherosclerosis process in elderly patients. J. Gerontol. A Biol. Sci. Med. Sci. 63, 200–203 (2008).
Chin, J. H., Okazaki, M., Hu, Z. W., Miller, J. W. & Hoffman, B. B. Activation of heat shock protein (hsp)70 and proto-oncogene expression by alpha1 adrenergic agonist in rat aorta with age. J. Clin. Invest. 97, 2316–2323 (1996).
Degenhardt, K. et al. Medin aggregation causes cerebrovascular dysfunction in aging wild-type mice. Proc. Natl Acad. Sci. USA 117, 23925–23931 (2020).
Mucchiano, G., Cornwell, G. G. & Westermark, P. Senile aortic amyloid. Evidence for two distinct forms of localized deposits. Am. J. Pathol. 140, 871–877 (1992).
Wagner, J. et al. Medin co-aggregates with vascular amyloid-β in Alzheimer’s disease. Nature 612, 123–131 (2022).
Migrino, R. Q. et al. Cerebrovascular medin is associated with Alzheimer’s disease and vascular dementia. Alzheimers Dement. 12, e12072 (2020).
Rajasekaran, N. S. et al. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 130, 427–439 (2007).
Tannous, P. et al. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc. Natl Acad. Sci. USA 105, 9745–9750 (2008).
Ma, X. et al. Transcription factor EB activation rescues advanced αB-crystallin mutation-induced cardiomyopathy by normalizing desmin localization. J. Am. Heart Assoc. 8, e010866 (2019).
Vicart, P. et al. A missense mutation in the αB-crystallin chaperone gene causes a desmin-related myopathy. Nat. Genet. 20, 92–95 (1998).
Nikolova, V. et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J. Clin. Invest. 113, 357–369 (2004).
Frock, R. L. et al. Cardiomyocyte-specific expression of lamin a improves cardiac function in Lmna−/− mice. PLoS One 7, e42918 (2012).
Galata, Z. et al. Amelioration of desmin network defects by αB-crystallin overexpression confers cardioprotection in a mouse model of dilated cardiomyopathy caused by LMNA gene mutation. J. Mol. Cell Cardiol. 125, 73–86 (2018).
Gupta, M. K. et al. Sumo E2 enzyme UBC9 is required for efficient protein quality control in cardiomyocytes. Circ. Res. 115, 721–729 (2014).
Pattison, J. S. et al. Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation 117, 2743–2751 (2008).
Blice-Baum, A. C. et al. Modest overexpression of FOXO maintains cardiac proteostasis and ameliorates age-associated functional decline. Aging Cell 16, 93–103 (2017).
Sanbe, A. et al. Protective effect of geranylgeranylacetone via enhancement of HSPB8 induction in desmin-related cardiomyopathy. PLoS One 4, e5351 (2009).
Ooie, T. et al. Single oral dose of geranylgeranylacetone induces heat-shock protein 72 and renders protection against ischemia/reperfusion injury in rat heart. Circulation 104, 1837–1843 (2001).
Elliott, P. et al. Long-term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Circ. Heart Fail. 15, e008193 (2022).
Maurer, M. S. et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 379, 1007–1016 (2018).
Fontana, L. Interventions to promote cardiometabolic health and slow cardiovascular ageing. Nat. Rev. Cardiol. 15, 566–577 (2018).
Yang, L. et al. Long-term calorie restriction enhances cellular quality-control processes in human skeletal muscle. Cell Rep. 14, 422–428 (2016).
Ono, T. et al. Age-associated increase of spontaneous mutant frequency and molecular nature of mutation in newborn and old lacZ-transgenic mouse. Mutat. Res. 447, 165–177 (2000).
De Majo, F. et al. Genomic instability in the naturally and prematurely aged myocardium. Proc. Natl Acad. Sci. USA 118, e2022974118 (2021).
Dollé, M. E., Snyder, W. K., Gossen, J. A., Lohman, P. H. & Vijg, J. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc. Natl Acad. Sci. USA 97, 8403–8408 (2000).
Busuttil, R. A., Dollé, M., Campisi, J. & Vijga, J. Genomic instability, aging, and cellular senescence. Ann. N. Y. Acad. Sci. 1019, 245–255 (2004).
Oh, H. et al. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc. Natl Acad. Sci. USA 98, 10308–10313 (2001).
Terai, M. et al. Association of telomere shortening in myocardium with heart weight gain and cause of death. Sci. Rep. 3, 2401 (2013).
Takubo, K. et al. Telomere lengths are characteristic in each human individual. Exp. Gerontol. 37, 523–531 (2002).
Anderson, R. et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 38, e100492 (2019).
Muñoz-Lorente, M. A., Cano-Martin, A. C. & Blasco, M. A. Mice with hyper-long telomeres show less metabolic aging and longer lifespans. Nat. Commun. 10, 4723 (2019).
Sharifi‐Sanjani, M. et al. Cardiomyocyte‐specific telomere shortening is a distinct signature of heart failure in humans. J. Am. Heart Assoc. 6, e005086 (2017).
Chang, A. C. Y. et al. Telomere shortening is a hallmark of genetic cardiomyopathies. Proc. Natl Acad. Sci. USA 115, 9276–9281 (2018).
Ungvari, Z., Tarantini, S., Donato, A. J., Galvan, V. & Csiszar, A. Mechanisms of vascular aging. Circ. Res. 123, 849–867 (2018).
Bloom, S. I. et al. Aging results in DNA damage and telomere dysfunction that is greater in endothelial versus vascular smooth muscle cells and is exacerbated in atheroprone regions. Geroscience 44, 2741–2755 (2022).
Martinet, W., Knaapen, M. W. M., De Meyer, G. R. Y., Herman, A. G. & Kockx, M. M. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 106, 927–932 (2002).
Gray, K. et al. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ. Res. 116, 816–826 (2015).
Yabluchanskiy, A. et al. Pharmacological or genetic depletion of senescent astrocytes prevents whole brain irradiation-induced impairment of neurovascular coupling responses protecting cognitive function in mice. Geroscience 42, 409–428 (2020).
Remes, T. M. et al. Radiation-induced accelerated aging of the brain vasculature in young adult survivors of childhood brain tumors. Neurooncol. Pract. 7, 415–427 (2020).
Andreassi, M. G. et al. Subclinical carotid atherosclerosis and early vascular aging from long-term low-dose ionizing radiation exposure: a genetic, telomere, and vascular ultrasound study in cardiac catheterization laboratory staff. JACC Cardiovasc. Interv. 8, 616–627 (2015).
Durik, M. et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 126, 468–478 (2012).
Ataei Ataabadi, E. et al. Vascular ageing features caused by selective DNA damage in smooth muscle cell. Oxid. Med. Cell Longev. 2021, 2308317 (2021).
Bautista-Niño, P. K. et al. Local endothelial DNA repair deficiency causes aging-resembling endothelial-specific dysfunction. Clin. Sci. 134, 727–746 (2020).
Matsumoto, T. et al. Aging-associated vascular phenotype in mutant mice with low levels of BubR1. Stroke 38, 1050–1056 (2007).
Jaiswal, S. & Libby, P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol. 17, 137–144 (2020).
Abelson, S. et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 559, 400–404 (2018).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Genovese, G. et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014).
Steensma, D. P. et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015).
Xie, M. et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 20, 1472–1478 (2014).
Jaiswal, S. et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 377, 111–121 (2017).
Dorsheimer, L. et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. 4, 25–33 (2019).
Shi, C. et al. Clonal haematopoiesis of indeterminate potential: associations with heart failure incidence, clinical parameters and biomarkers. Eur. J. Heart Fail. 25, 4–13 (2023).
Devesa, A. et al. Bone marrow activation in response to metabolic syndrome and early atherosclerosis. Eur. Heart J. 43, 1809–1828 (2022).
Schloss, M. J., Swirski, F. K. & Nahrendorf, M. Modifiable cardiovascular risk, hematopoiesis, and innate immunity. Circ. Res. 126, 1242–1259 (2020).
Fuster, J. J. et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017).
Sano, S. et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol. 71, 875–886 (2018).
Sano, S. et al. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ. Res. 123, 335–341 (2018).
Zekavat, S. M. et al. TP53-mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease. Nat. Cardiovasc. Res. 2, 144–158 (2023).
Abplanalp, W. T. et al. Clonal hematopoiesis-driver DNMT3A mutations alter immune cells in heart failure. Circ. Res. 128, 216–228 (2021).
Bick, A. G. et al. Genetic Interleukin 6 signaling deficiency attenuates cardiovascular risk in clonal hematopoiesis. Circulation 141, 124–131 (2020).
Kabir, I. et al. The age of bone marrow dictates the clonality of smooth muscle-derived cells in atherosclerotic plaques. Nat. Aging 3, 64–81 (2023).
Potus, F. et al. Novel mutations and decreased expression of the epigenetic regulator TET2 in pulmonary arterial hypertension. Circulation 141, 1986–2000 (2020).
Shi, Y. et al. Epigenetic regulation in cardiovascular disease: mechanisms and advances in clinical trials. Signal. Transduct. Target. Ther. 7, 200 (2022).
Greco, C. M. & Condorelli, G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat. Rev. Cardiol. 12, 488–497 (2015).
Schaum, N. et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 583, 596–602 (2020).
Stoeger, T. et al. Aging is associated with a systemic length-associated transcriptome imbalance. Nat. Aging 2, 1191–1206 (2022).
Bahar, R. et al. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441, 1011–1014 (2006).
Jusic, A. et al. Noncoding RNAs in age-related cardiovascular diseases. Ageing Res. Rev. 77, 101610 (2022).
Du, W. W. et al. Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur. Heart J. 38, 1402–1412 (2017).
Zhang, X., Azhar, G. & Wei, J. Y. The expression of microRNA and microRNA clusters in the aging heart. PLoS One 7, e34688 (2012).
Boon, R. A. et al. MicroRNA-34a regulates cardiac ageing and function. Nature 495, 107–110 (2013).
Ito, T., Yagi, S. & Yamakuchi, M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun. 398, 735–740 (2010).
Badi, I. et al. MicroRNA-34a induces vascular smooth muscle cells senescence by SIRT1 downregulation and promotes the expression of age-associated pro-inflammatory secretory factors. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1304–1311 (2015).
Badi, I. et al. miR-34a promotes vascular smooth muscle cell calcification by downregulating SIRT1 (Sirtuin 1) and Axl (AXL receptor tyrosine kinase). Arterioscler. Thromb. Vasc. Biol. 38, 2079–2090 (2018).
Huang, Z.-P. et al. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ. Res. 112, 1234–1243 (2013).
Gupta, S. K. et al. Preclinical development of a microRNA-based therapy for elderly patients with myocardial infarction. J. Am. Coll. Cardiol. 68, 1557–1571 (2016).
Mattick, J. S. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-022-00566-8 (2023).
Ponting, C. P. & Haerty, W. Genome-wide analysis of human long noncoding RNAs: a provocative review. Annu. Rev. Genomics Hum. Genet. 23, 153–172 (2022).
Trembinski, D. J. et al. Aging-regulated anti-apoptotic long non-coding RNA Sarrah augments recovery from acute myocardial infarction. Nat. Commun. 11, 2039 (2020).
Lu, T.-M. et al. Downregulation of Sirt1 as aging change in advanced heart failure. J. Biomed. Sci. 21, 57 (2014).
Donato, A. J. et al. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 589, 4545–4554 (2011).
Alcendor, R. R. et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100, 1512–1521 (2007).
Guo, Y. et al. Endothelial SIRT1 prevents age-induced impairment of vasodilator responses by enhancing the expression and activity of soluble guanylyl cyclase in smooth muscle cells. Cardiovasc. Res. 115, 678–690 (2019).
Xu, S. et al. SIRT6 protects against endothelial dysfunction and atherosclerosis in mice. Aging 8, 1064–1082 (2016).
Li, W. et al. SIRT6 protects vascular smooth muscle cells from osteogenic transdifferentiation via Runx2 in chronic kidney disease. J. Clin. Invest. 132, e150051 (2022).
Sundaresan, N. R. et al. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat. Med. 18, 1643–1650 (2012).
Zhang, W., Song, M., Qu, J. & Liu, G.-H. Epigenetic modifications in cardiovascular aging and diseases. Circ. Res. 123, 773–786 (2018).
Jeong, M. Y. et al. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci. Transl. Med. 10, eaao0144 (2018).
Travers, J. G. et al. HDAC inhibition reverses preexisting diastolic dysfunction and blocks covert extracellular matrix remodeling. Circulation 143, 1874–1890 (2021).
Wallner, M. et al. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci. Transl. Med. 12, eaay7205 (2020).
Bagchi, R. A. & Weeks, K. L. Histone deacetylases in cardiovascular and metabolic diseases. J. Mol. Cell Cardiol. 130, 151–159 (2019).
Demos-Davies, K. M. et al. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am. J. Physiol. Heart Circ. Physiol. 307, H252–H258 (2014).
Boucherat, O. et al. HDAC6: a novel histone deacetylase implicated in pulmonary arterial hypertension. Sci. Rep. 7, 4546 (2017).
Lin, Y.-H. et al. HDAC6 modulates myofibril stiffness and diastolic function of the heart. J. Clin. Invest. 132, e148333 (2022).
Malhotra, R. et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 51, 1580–1587 (2019).
Zhang, C. L. et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488 (2002).
Nguyen, A. T. et al. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 25, 263–274 (2011).
Stein, A. B. et al. Loss of H3K4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J. Clin. Invest. 121, 2641–2650 (2011).
Delgado-Olguín, P. et al. Epigenetic repression of cardiac progenitor gene expression by Ezh2 is required for postnatal cardiac homeostasis. Nat. Genet. 44, 343–347 (2012).
Papait, R. et al. Histone methyltransferase G9a is required for cardiomyocyte homeostasis and hypertrophy. Circulation 136, 1233–1246 (2017).
Farina, F. M. et al. The epigenetic enzyme DOT1L orchestrates vascular smooth muscle cell-monocyte crosstalk and protects against atherosclerosis via the NF-κB pathway. Eur. Heart J. 43, 4562–4576 (2022).
Thienpont, B. et al. The H3K9 dimethyltransferases EHMT1/2 protect against pathological cardiac hypertrophy. J. Clin. Invest. 127, 335–348 (2017).
Lind, L., Ingelsson, E., Sundström, J., Siegbahn, A. & Lampa, E. Methylation-based estimated biological age and cardiovascular disease. Eur. J. Clin. Invest. 48, 12872 (2018).
Lo, Y.-H. & Lin, W.-Y. Cardiovascular health and four epigenetic clocks. Clin. Epigenetics 14, 73 (2022).
Zheng, Y. et al. Association of cardiovascular health through young adulthood with genome-wide DNA methylation patterns in midlife: the CARDIA study. Circulation 146, 94–109 (2022).
Movassagh, M. et al. Distinct epigenomic features in end-stage failing human hearts. Circulation 124, 2411–2422 (2011).
Haas, J. et al. Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 5, 413–429 (2013).
Pepin, M. E. et al. DNA methylation reprograms cardiac metabolic gene expression in end-stage human heart failure. Am. J. Physiol. Heart Circ. Physiol. 317, H674–H684 (2019).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Pavanello, S. et al. The biological age of the heart is consistently younger than chronological age. Sci. Rep. 10, 10752 (2020).
Wu, T.-T. et al. Myocardial tissue-specific Dnmt1 knockout in rats protects against pathological injury induced by adriamycin. Lab. Invest. 100, 974–985 (2020).
Nührenberg, T. G. et al. Cardiac myocyte de novo DNA methyltransferases 3a/3b are dispensable for cardiac function and remodeling after chronic pressure overload in mice. PLoS One 10, e0131019 (2015).
Vujic, A. et al. Experimental heart failure modelled by the cardiomyocyte-specific loss of an epigenome modifier, DNMT3B. J. Mol. Cell Cardiol. 82, 174–183 (2015).
Okano, M., Bell, D. W., Haber, D. A. & Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257 (1999).
Tyrrell, D. J. & Goldstein, D. R. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat. Rev. Cardiol. 18, 58–68 (2021).
Heyn, H. et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl Acad. Sci. USA 109, 10522–10527 (2012).
Zaina, S. et al. DNA methylation map of human atherosclerosis. Circ. Cardiovasc. Genet. 7, 692–700 (2014).
Dunn, J. et al. Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J. Clin. Invest. 124, 3187–3199 (2014).
Amorim, J. A. et al. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 18, 243–258 (2022).
Wallace, D. C. A mitochondrial bioenergetic etiology of disease. J. Clin. Invest. 123, 1405–1412 (2013).
Galluzzi, L., Kepp, O., Trojel-Hansen, C. & Kroemer, G. Mitochondrial control of cellular life, stress, and death. Circ. Res. 111, 1198–1207 (2012).
Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T. & Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 23, 159–173 (2023).
Lopez-Crisosto, C. et al. Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 14, 342–360 (2017).
Lesnefsky, E. J., Chen, Q. & Hoppel, C. L. Mitochondrial metabolism in aging heart. Circ. Res. 118, 1593–1611 (2016).
Dai, D.-F., Rabinovitch, P. S. & Ungvari, Z. Mitochondria and cardiovascular aging. Circ. Res. 110, 1109–1124 (2012).
Tian, R. et al. Unlocking the secrets of mitochondria in the cardiovascular system: path to a cure in heart failure — a report from the 2018 National Heart, Lung, and Blood Institute Workshop. Circulation 140, 1205–1216 (2019).
Bonora, M. et al. Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat. Rev. Cardiol. 16, 33–55 (2019).
Fannin, S. W., Lesnefsky, E. J., Slabe, T. J., Hassan, M. O. & Hoppel, C. L. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch. Biochem. Biophys. 372, 399–407 (1999).
Tatarková, Z. et al. Effects of aging on activities of mitochondrial electron transport chain complexes and oxidative damage in rat heart. Physiol. Res. 60, 281–289 (2011).
Lesnefsky, E. J. et al. Aging decreases electron transport complex III activity in heart interfibrillar mitochondria by alteration of the cytochrome c binding site. J. Mol. Cell Cardiol. 33, 37–47 (2001).
Messerer, J. et al. Spermidine supplementation influences mitochondrial number and morphology in the heart of aged mice. J. Anat. 242, 91–101 (2023).
Kates, A. M. et al. Impact of aging on substrate metabolism by the human heart. J. Am. Coll. Cardiol. 41, 293–299 (2003).
McMillin, J. B., Taffet, G. E., Taegtmeyer, H., Hudson, E. K. & Tate, C. A. Mitochondrial metabolism and substrate competition in the aging Fischer rat heart. Cardiovasc. Res. 27, 2222–2228 (1993).
Ungvari, Z. et al. Dysregulation of mitochondrial biogenesis in vascular endothelial and smooth muscle cells of aged rats. Am. J. Physiol. Heart Circ. Physiol. 294, H2121–H2128 (2008).
Foote, K. et al. Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell 17, e12773 (2018).
Park, S.-Y. et al. Age-related endothelial dysfunction in human skeletal muscle feed arteries: the role of free radicals derived from mitochondria in the vasculature. Acta Physiol. 222, e12893 (2018).
Gioscia-Ryan, R. A. et al. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 592, 2549–2561 (2014).
Ungvari, Z. et al. Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 301, H363–H372 (2011).
Ungvari, Z. et al. Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-κB activation in the nonhuman primate Macaca mulatta. J. Gerontol. A Biol. Sci. Med. Sci. 66, 866–875 (2011).
Tarantini, S. et al. Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood-brain barrier disruption, neuroinflammation, amyloidogenic gene expression, and cognitive decline in mice, mimicking the aging phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 73, 853–863 (2018).
Fulop, G. A. et al. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience 40, 513–521 (2018).
Ungvari, Z. et al. Increased mitochondrial H2O2 production promotes endothelial NF-κB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 293, H37–H47 (2007).
Yu, H. et al. Role of the cGAS-STING pathway in aging-related endothelial dysfunction. Aging Dis. 13, 1901 (2022).
Rech, L. & Rainer, P. P. The innate immune cGAS-STING-pathway in cardiovascular diseases — a mini review. Front. Cardiovasc. Med. 8, 715903 (2021).
Pierce, G. L., Lesniewski, L. A., Lawson, B. R., Beske, S. D. & Seals, D. R. Nuclear factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation 119, 1284–1292 (2009).
Tyrrell, D. J. et al. Age-associated mitochondrial dysfunction accelerates atherogenesis. Circ. Res. 126, 298–314 (2020).
Abdellatif, M., Sedej, S. & Kroemer, G. NAD+ metabolism in cardiac health, aging, and disease. Circulation 144, 1795–1817 (2021).
Abdellatif, M., Bugger, H., Kroemer, G. & Sedej, S. NAD+ and vascular dysfunction: from mechanisms to therapeutic opportunities. J. Lipid Atheroscler. 11, 111–132 (2022).
Zimmermann, A., Madreiter-Sokolowski, C., Stryeck, S. & Abdellatif, M. Targeting the mitochondria-proteostasis axis to delay aging. Front. Cell Dev. Biol. 9, 656201 (2021).
Hoshino, A. et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 4, 2308 (2013).
Kubli, D. A., Quinsay, M. N. & Gustafsson, A. B. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun. Integr. Biol. 6, e24511 (2013).
Diao, R. Y. & Gustafsson, Å. B. Mitochondrial quality surveillance: mitophagy in cardiovascular health and disease. Am. J. Physiol. Cell Physiol. 322, C218–C230 (2022).
Billia, F. et al. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc. Natl Acad. Sci. USA 108, 9572–9577 (2011).
Zhu, X. et al. Fine-tuning of PGC1α expression regulates cardiac function and longevity. Circ. Res. 125, 707–719 (2019).
Song, M., Franco, A., Fleischer, J. A., Zhang, L. & Dorn, G. W. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. 26, 872–883.e5 (2017).
Wang, Q. et al. Deletion of PRKAA triggers mitochondrial fission by inhibiting the autophagy-dependent degradation of DNM1L. Autophagy 13, 404–422 (2017).
Peoples, J. N., Saraf, A., Ghazal, N., Pham, T. T. & Kwong, J. Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 51, 1–13 (2019).
Dai, D.-F. et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 9, 536–544 (2010).
Trifunovic, A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004).
Yu, E. et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 128, 702–712 (2013).
Shabalina, I. G. et al. Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1. Aging 9, 315–339 (2017).
Fernandez-Caggiano, M. et al. Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nat. Metab. 2, 1223–1231 (2020).
Zhang, Y. et al. Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nat. Metab. 2, 1248–1264 (2020).
De Bock, K. et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154, 651–663 (2013).
Wilson, C., Lee, M. D., Buckley, C., Zhang, X. & McCarron, J. G. Mitochondrial ATP production is required for endothelial cell control of vascular tone. Function 4, zqac063 (2022).
Xiong, J. et al. A metabolic basis for endothelial-to-mesenchymal transition. Mol. Cell 69, 689–698.e7 (2018).
Tarantini, S. et al. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell 17, e12731 (2018).
Rossman, M. J. et al. Chronic supplementation with a mitochondrial antioxidant (MitoQ) improves vascular function in healthy older adults. Hypertension 71, 1056–1063 (2018).
Schriner, S. E. et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308, 1909–1911 (2005).
Yu, E. P. K. et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler. Thromb. Vasc. Biol. 37, 2322–2332 (2017).
Kataura, T. et al. Autophagy promotes cell survival by maintaining NAD levels. Dev. Cell 57, 2584–2598.e11 (2022).
Abdellatif, M. et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci. Transl. Med. 13, eabd7064 (2021).
de Picciotto, N. E. et al. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15, 522–530 (2016).
Tarantini, S. et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 24, 101192 (2019).
Das, A. et al. Impairment of an endothelial NAD+-H2S signaling network is a reversible cause of vascular aging. Cell 173, 74–89.e20 (2018).
Kiss, T. et al. Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. Geroscience 42, 527–546 (2020).
Tarantini, S. et al. Treatment with the poly(ADP-ribose) polymerase inhibitor PJ-34 improves cerebromicrovascular endothelial function, neurovascular coupling responses and cognitive performance in aged mice, supporting the NAD+ depletion hypothesis of neurovascular aging. Geroscience 41, 533–542 (2019).
Mehdizadeh, M., Aguilar, M., Thorin, E., Ferbeyre, G. & Nattel, S. The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat. Rev. Cardiol. 19, 250–264 (2022).
Gude, N. A., Broughton, K. M., Firouzi, F. & Sussman, M. A. Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nat. Rev. Cardiol. 15, 523–542 (2018).
Gorgoulis, V. et al. Cellular senescence: defining a path forward. Cell 179, 813–827 (2019).
Di Micco, R., Krizhanovsky, V., Baker, D. & d’Adda di Fagagna, F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 22, 75–95 (2021).
Admasu, T. D., Rae, M. & Stolzing, A. Dissecting primary and secondary senescence to enable new senotherapeutic strategies. Ageing Res. Rev. 70, 101412 (2021).
González-Gualda, E., Baker, A. G., Fruk, L. & Muñoz-Espín, D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 288, 56–80 (2021).
Kohli, J. et al. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat. Protoc. 16, 2471–2498 (2021).
Yousefzadeh, M. J. et al. An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105 (2021).
Owens, W. A., Walaszczyk, A., Spyridopoulos, I., Dookun, E. & Richardson, G. D. Senescence and senolytics in cardiovascular disease: promise and potential pitfalls. Mech. Ageing Dev. 198, 111540 (2021).
Honda, S. et al. Cellular senescence promotes endothelial activation through epigenetic alteration, and consequently accelerates atherosclerosis. Sci. Rep. 11, 14608 (2021).
Wang, J. et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation 132, 1909–1919 (2015).
Childs, B. G. et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477 (2016).
Dookun, E. et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 19, e13249 (2020).
Gevaert, A. B. et al. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ. Heart Fail. 10, e003806 (2017).
Moiseeva, V. et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature 613, 169–178 (2023).
Bloom, S. I., Islam, M. T., Lesniewski, L. A. & Donato, A. J. Mechanisms and consequences of endothelial cell senescence. Nat. Rev. Cardiol. 20, 38–51 (2023).
Grosse, L. et al. Defined p16High senescent cell types are indispensable for mouse healthspan. Cell Metab. 32, 87–99.e6 (2020).
Kiss, T. et al. Single-cell RNA sequencing identifies senescent cerebromicrovascular endothelial cells in the aged mouse brain. Geroscience 42, 429–444 (2020).
Chala, N. et al. Mechanical fingerprint of senescence in endothelial cells. Nano Lett. 21, 4911–4920 (2021).
Bhayadia, R., Schmidt, B. M. W., Melk, A. & Hömme, M. Senescence-induced oxidative stress causes endothelial dysfunction. J. Gerontol. A Biol. Sci. Med. Sci. 71, 161–169 (2016).
Yamazaki, Y. et al. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke 47, 1068–1077 (2016).
Baker, D. J. et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
Meyer, K., Hodwin, B., Ramanujam, D., Engelhardt, S. & Sarikas, A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J. Am. Coll. Cardiol. 67, 2018–2028 (2016).
Garrido, A. M. et al. Efficacy and limitations of senolysis in atherosclerosis. Cardiovasc. Res. 118, 1713–1727 (2022).
Gasek, N. S., Kuchel, G. A., Kirkland, J. L. & Xu, M. Strategies for targeting senescent cells in human disease. Nat. Aging 1, 870–879 (2021).
Xu, M. et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24, 1246–1256 (2018).
Roos, C. M. et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15, 973–977 (2016).
Tarantini, S. et al. Treatment with the BCL-2/BCL-xL inhibitor senolytic drug ABT263/Navitoclax improves functional hyperemia in aged mice. Geroscience 43, 2427–2440 (2021).
Morgan, R. G. et al. Induced Trf2 deletion leads to aging vascular phenotype in mice associated with arterial telomere uncapping, senescence signaling, and oxidative stress. J. Mol. Cell Cardiol. 127, 74–82 (2019).
Leri, A. et al. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 22, 131–139 (2003).
Rossman, M. J. et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am. J. Physiol. Heart Circ. Physiol. 313, H890–H895 (2017).
Ziff, O. J. & Kotecha, D. Digoxin: the good and the bad. Trends Cardiovasc. Med. 26, 585–595 (2016).
Triana-Martínez, F. et al. Identification and characterization of cardiac glycosides as senolytic compounds. Nat. Commun. 10, 4731 (2019).
Ma, T. K. W., Kam, K. K. H., Yan, B. P. & Lam, Y.-Y. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. Br. J. Pharmacol. 160, 1273–1292 (2010).
Ruggenenti, P. et al. Renal and systemic effects of calorie restriction in patients with type 2 diabetes with abdominal obesity: a randomized controlled trial. Diabetes 66, 75–86 (2017).
Jia, G., Aroor, A. R., Hill, M. A. & Sowers, J. R. Role of renin-angiotensin-aldosterone system activation in promoting cardiovascular fibrosis and stiffness. Hypertension 72, 537–548 (2018).
Conti, S., Cassis, P. & Benigni, A. Aging and the renin-angiotensin system. Hypertension 60, 878–883 (2012).
Yoon, H. E. et al. Age-associated changes in the vascular renin-angiotensin system in mice. Oxid. Med. Cell Longev. 2016, 6731093 (2016).
Dai, D.-F. et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 119, 2789–2797 (2009).
Dinh, Q. N. et al. Pressor response to angiotensin II is enhanced in aged mice and associated with inflammation, vasoconstriction and oxidative stress. Aging 9, 1595–1606 (2017).
Forrester, S. J. et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol. Rev. 98, 1627–1738 (2018).
Billet, S. et al. Gain-of-function mutant of angiotensin II receptor, type 1A, causes hypertension and cardiovascular fibrosis in mice. J. Clin. Invest. 117, 1914–1925 (2007).
Habibi, J. et al. Mineralocorticoid receptor blockade improves diastolic function independent of blood pressure reduction in a transgenic model of RAAS overexpression. Am. J. Physiol. Heart Circ. Physiol. 300, H1484–H1491 (2011).
Xiao, H. D. et al. Mice with cardiac-restricted angiotensin-converting enzyme (ACE) have atrial enlargement, cardiac arrhythmia, and sudden death. Am. J. Pathol. 165, 1019–1032 (2004).
Benigni, A. et al. Disruption of the Ang II type 1 receptor promotes longevity in mice. J. Clin. Invest. 119, 524–530 (2009).
Basso, N. et al. Protective effect of long-term angiotensin II inhibition. Am. J. Physiol. Heart Circ. Physiol. 293, H1351–H1358 (2007).
Inserra, F., Romano, L., Ercole, L., de Cavanagh, E. M. & Ferder, L. Cardiovascular changes by long-term inhibition of the renin-angiotensin system in aging. Hypertension 25, 437–442 (1995).
Keller, K., Kane, A., Heinze-Milne, S., Grandy, S. A. & Howlett, S. E. Chronic treatment with the ACE inhibitor enalapril attenuates the development of frailty and differentially modifies pro- and anti-inflammatory cytokines in aging male and female C57BL/6 mice. J. Gerontol. A Biol. Sci. Med. Sci. 74, 1149–1157 (2019).
McCurley, A. et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat. Med. 18, 1429–1433 (2012).
Paz Ocaranza, M. et al. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 17, 116–129 (2020).
Lymperopoulos, A., Rengo, G. & Koch, W. J. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ. Res. 113, 739–753 (2013).
Elia, A. et al. Aging is associated with cardiac autonomic nerve fiber depletion and reduced cardiac and circulating BDNF levels. J. Geriatr. Cardiol. 18, 549–559 (2021).
Ferrara, N. et al. β-Adrenergic receptor responsiveness in aging heart and clinical implications. Front. Physiol. 4, 396 (2014).
Stratton, J. R. et al. Differences in cardiovascular responses to isoproterenol in relation to age and exercise training in healthy men. Circulation 86, 504–512 (1992).
Xiao, R. P., Spurgeon, H. A., O’Connor, F. & Lakatta, E. G. Age-associated changes in β-adrenergic modulation on rat cardiac excitation-contraction coupling. J. Clin. Invest. 94, 2051–2059 (1994).
Engelhardt, S., Hein, L., Wiesmann, F. & Lohse, M. J. Progressive hypertrophy and heart failure in β1-adrenergic receptor transgenic mice. Proc. Natl Acad. Sci. USA 96, 7059–7064 (1999).
Milano, C. A. et al. Enhanced myocardial function in transgenic mice overexpressing the β2-adrenergic receptor. Science 264, 582–586 (1994).
Du, X. Age-dependent cardiomyopathy and heart failure phenotype in mice overexpressing β2-adrenergic receptors in the heart. Cardiovasc. Res. 48, 448–454 (2000).
Bisognano, J. D. et al. Myocardial-directed overexpression of the human β1-adrenergic receptor in transgenic mice. J. Mol. Cell Cardiol. 32, 817–830 (2000).
Liggett, S. B. et al. Early and delayed consequences of β2-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation 101, 1707–1714 (2000).
No authors listed. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised intervention trial in congestive heart failure (MERIT-HF). Lancet 353, 2001–2007 (1999).
No authors listed. The cardiac insufficiency bisoprolol study II (CIBIS-II): a randomised trial. Lancet 353, 9–13 (1999).
Ziff, O. J. et al. Beta-blocker efficacy across different cardiovascular indications: an umbrella review and meta-analytic assessment. BMC Med. 18, 103 (2020).
Sipahi, I. et al. β-Blockers and progression of coronary atherosclerosis: pooled analysis of 4 intravascular ultrasonography trials. Ann. Intern. Med. 147, 10–18 (2007).
Leosco, D. et al. Exercise training and β-blocker treatment ameliorate age-dependent impairment of β-adrenergic receptor signaling and enhance cardiac responsiveness to adrenergic stimulation. Am. J. Physiol. Heart Circ. Physiol. 293, H1596–H1603 (2007).
Spindler, S. R. et al. β1-Adrenergic receptor blockade extends the life span of Drosophila and long-lived mice. Age 35, 2099–2109 (2013).
Yan, L. et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell 130, 247–258 (2007).
del Villar, S. G., Westhoff, M., Voelker, T. L., Dickson, E. J. & Dixon, R. E. Restoration of β-adrenergic responsivity in the aging heart. Preprint at bioRxiv https://doi.org/10.1101/2022.08.23.504979 (2022).
Vögele, A. et al. Effectiveness and safety of beta blockers in the management of hypertension in older adults: a systematic review to help reduce inappropriate prescribing. BMC Geriatr. 17, 224 (2017).
Chan You, S. et al. Comprehensive comparative effectiveness and safety of first-line β-blocker monotherapy in hypertensive patients: a large-scale multicenter observational study. Hypertension 77, 1528–1538 (2021).
Junnila, R. K., List, E. O., Berryman, D. E., Murrey, J. W. & Kopchick, J. J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 9, 366–376 (2013).
Milman, S., Huffman, D. M. & Barzilai, N. The somatotropic axis in human aging: framework for the current state of knowledge and future research. Cell Metab. 23, 980–989 (2016).
McMullen, J. R. et al. The insulin-like growth factor 1 receptor induces physiological heart growth via the phosphoinositide 3-kinase(p110α) pathway. J. Biol. Chem. 279, 4782–4793 (2004).
Delaughter, M. C., Taffet, G. E., Fiorotto, M. L., Entman, M. L. & Schwartz, R. J. Local insulin-like growth factor I expression induces physiologic, then pathologic, cardiac hypertrophy in transgenic mice. FASEB J. 13, 1923–1929 (1999).
Ock, S. et al. Deletion of IGF-1 receptors in cardiomyocytes attenuates cardiac aging in male mice. Endocrinology 157, 336–345 (2016).
Mao, K. et al. Late-life targeting of the IGF-1 receptor improves healthspan and lifespan in female mice. Nat. Commun. 9, 2394 (2018).
Inuzuka, Y. et al. Suppression of phosphoinositide 3-kinase prevents cardiac aging in mice. Circulation 120, 1695–1703 (2009).
Hedges, C. P. et al. Dietary supplementation of clinically utilized PI3K p110α inhibitor extends the lifespan of male and female mice. Nat. Aging 3, 162–172 (2023).
D’Assante, R. et al. Myocardial expression of somatotropic axis, adrenergic signalling, and calcium handling genes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. Esc. Heart Fail. 8, 1681–1686 (2021).
Vasan, R. S. et al. Serum insulin-like growth factor I and risk for heart failure in elderly individuals without a previous myocardial infarction: the Framingham Heart Study. Ann. Intern. Med. 139, 642–648 (2003).
Andreassen, M. et al. IGF1 as predictor of all cause mortality and cardiovascular disease in an elderly population. Eur. J. Endocrinol. 160, 25–31 (2009).
Rahmani, J. et al. Association between IGF-1 levels ranges and all-cause mortality: a meta-analysis. Aging Cell 21, e13540 (2022).
Zhang, W. B., Ye, K., Barzilai, N. & Milman, S. The antagonistic pleiotropy of insulin-like growth factor 1. Aging Cell 20, e13443 (2021).
Fontana, L. et al. Identification of a metabolic signature for multidimensional impairment and mortality risk in hospitalized older patients. Aging Cell 12, 459–466 (2013).
Toth, L. et al. Age-related decline in circulating IGF-1 associates with impaired neurovascular coupling responses in older adults. Geroscience 44, 2771–2783 (2022).
Erlandsson, M. C. et al. Low serum IGF1 is associated with hypertension and predicts early cardiovascular events in women with rheumatoid arthritis. BMC Med. 17, 141 (2019).
Csiszar, A. et al. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. Am. J. Physiol. Heart Circ. Physiol. 295, H1882–H1894 (2008).
Reddy, A. K. et al. Young little mice express a premature cardiovascular aging phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 69, 152–159 (2014).
Abbas, A. et al. The insulin-like growth factor-1 receptor is a negative regulator of nitric oxide bioavailability and insulin sensitivity in the endothelium. Diabetes 60, 2169–2178 (2011).
Higashi, Y. et al. Endothelial deficiency of insulin-like growth factor-1 receptor reduces endothelial barrier function and promotes atherosclerosis in Apoe-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 319, H730–H743 (2020).
Franceschi, C. et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254 (2000).
Ferrucci, L. & Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522 (2018).
Engelen, S. E., Robinson, A. J. B., Zurke, Y.-X. & Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat. Rev. Cardiol. 19, 522–542 (2022).
Libby, P., Ridker, P. M. & Maseri, A. Inflammation and atherosclerosis. Circulation 105, 1135–1143 (2002).
Dutta, P. et al. Myocardial infarction accelerates atherosclerosis. Nature 487, 325–329 (2012).
Joshi, N. V. et al. Systemic atherosclerotic inflammation following acute myocardial infarction: myocardial infarction begets myocardial infarction. J. Am. Heart Assoc. 4, e001956 (2015).
Esfahani, N. S. et al. Aging influences the cardiac macrophage phenotype and function during steady state and during inflammation. Aging Cell 20, e13438 (2021).
Koenig, A. L. et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat. Cardiovasc. Res. 1, 263–280 (2022).
Barcena de Arellano, M. L. et al. Sex differences in the aging human heart: decreased sirtuins, pro-inflammatory shift and reduced anti-oxidative defense. Aging 11, 1918–1933 (2019).
Ramos, G. C. et al. Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl Acad. Sci. USA 114, E2420–E2429 (2017).
Delgobo, M. et al. Terminally differentiated CD4+ T cells promote myocardial inflammaging. Front. Immunol. 12, 584538 (2021).
Dolejsi, T. et al. Adult T-cells impair neonatal cardiac regeneration. Eur. Heart J. 43, 2698–2709 (2022).
Desdín-Micó, G. et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376 (2020).
Maier, H. J. et al. Cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc. Natl Acad. Sci. USA 109, 11794–11799 (2012).
Kubota, T. et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-α. Circ. Res. 81, 627–635 (1997).
Marín-Aguilar, F. et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell 19, e13050 (2020).
Hasegawa, Y. et al. Blockade of the nuclear factor-κB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation 125, 1122–1133 (2012).
Tardif, J.-C. et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 381, 2497–2505 (2019).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Everett, B. M. et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 139, 1289–1299 (2019).
Ridker, P. M. et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842 (2017).
Dunlay, S. M., Roger, V. L. & Redfield, M. M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 14, 591–602 (2017).
Paulus, W. J. & Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 62, 263–271 (2013).
Abdellatif, M. & Kroemer, G. Heart failure with preserved ejection fraction: an age-related condition. J. Mol. Cell Cardiol. 167, 83–84 (2022).
Withaar, C., Li, S., Meems, L. M. G., Silljé, H. H. W. & de Boer, R. A. Aging and HFpEF: are we running out of time? J. Mol. Cell Cardiol. 168, 33–34 (2022).
Kalogeropoulos, A. et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J. Am. Coll. Cardiol. 55, 2129–2137 (2010).
Solomon, S. D. et al. Dapagliflozin in heart failure with mildly reduced or preserved ejection fraction. N. Engl. J. Med. 387, 1089–1098 (2022).
Anker, S. D. et al. Empagliflozin in heart failure with a preserved ejection fraction. N. Engl. J. Med. 385, 1451–1461 (2021).
Elrakaybi, A., Laubner, K., Zhou, Q., Hug, M. J. & Seufert, J. Cardiovascular protection by SGLT2 inhibitors – Do anti-inflammatory mechanisms play a role? Mol. Metab. 64, 101549 (2022).
Cowie, M. R. & Fisher, M. SGLT2 inhibitors: mechanisms of cardiovascular benefit beyond glycaemic control. Nat. Rev. Cardiol. 17, 761–772 (2020).
Zannad, F. et al. Effect of empagliflozin on circulating proteomics in heart failure: mechanistic insights into the EMPEROR programme. Eur. Heart J. 43, 4991–5002 (2022).
Hoong, C. W. S. & Chua, M. W. J. SGLT2 Inhibitors as calorie restriction mimetics: insights on longevity pathways and age-related diseases. Endocrinology 162, bqab079 (2021).
Packer, M. SGLT2 inhibitors: role in protective reprogramming of cardiac nutrient transport and metabolism. Nat. Rev. Cardiol. https://doi.org/10.1038/s41569-022-00824-4 (2023).
Deng, Y. et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ. Res. 128, 232–245 (2021).
Yang, H., Wang, H., Ren, J., Chen, Q. & Chen, Z. J. cGAS is essential for cellular senescence. Proc. Natl Acad. Sci. USA 114, E4612–E4620 (2017).
Sladitschek-Martens, H. L. et al. YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature 607, 790–798 (2022).
Kreienkamp, R. et al. A cell-intrinsic interferon-like response links replication stress to cellular aging caused by progerin. Cell Rep. 22, 2006–2015 (2018).
Olive, M. et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler. Thromb. Vasc. Biol. 30, 2301–2309 (2010).
Pham, P. T. et al. STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur. Heart J. 42, 4336–4348 (2021).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487 (2017).
Cao, D. J. et al. Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 137, 2613–2634 (2018).
Li, Q. et al. Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol. Med. 12, e11002 (2020).
Rech, L. et al. Small molecule STING inhibition improves myocardial infarction remodeling. Life Sci. 291, 120263 (2022).
Mittelbrunn, M. & Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 22, 687–698 (2021).
Castoldi, F. et al. Autophagy-mediated metabolic effects of aspirin. Cell Death Discov. 6, 129 (2020).
McNeil, J. J. et al. Effect of aspirin on cardiovascular events and bleeding in the healthy elderly. N. Engl. J. Med. 379, 1509–1518 (2018).
Gaziano, J. M. et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): a randomised, double-blind, placebo-controlled trial. Lancet 392, 1036–1046 (2018).
ASCEND Study Collaborative Group et al. Effects of aspirin for primary prevention in persons with diabetes mellitus. N. Engl. J. Med. 379, 1529–1539 (2018).
Castellano, J. M. et al. Polypill strategy in secondary cardiovascular prevention. N. Engl. J. Med. 387, 967–977 (2022).
Svensson, E. C. et al. TET2-Driven clonal hematopoiesis and response to canakinumab: an exploratory analysis of the CANTOS randomized clinical trial. JAMA Cardiol. 7, 521–528 (2022).
Ridker, P. M. et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 391, 319–328 (2018).
Rajendran, P. et al. Autophagy and senescence: a new insight in selected human diseases. J. Cell Physiol. 234, 21485–21492 (2019).
Levine, B., Mizushima, N. & Virgin, H. W. Autophagy in immunity and inflammation. Nature 469, 323–335 (2011).
Jaiswal, S. & Ebert, B. L. Clonal hematopoiesis in human aging and disease. Science 366, eaan4673 (2019).
Powell-Wiley, T. M. et al. Obesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 143, e984–e1010 (2021).
Tong, M. et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ. Res. 124, 1360–1371 (2019).
Sánchez-Ceinos, J. et al. Impaired mRNA splicing and proteostasis in preadipocytes in obesity-related metabolic disease. eLife 10, e65996 (2021).
Park, Y. J., Han, S. M., Huh, J. Y. & Kim, J. B. Emerging roles of epigenetic regulation in obesity and metabolic disease. J. Biol. Chem. 297, 101296 (2021).
Abel, E. D. Obesity stresses cardiac mitochondria even when you are young. J. Am. Coll. Cardiol. 57, 586–589 (2011).
Chaib, S., Tchkonia, T. & Kirkland, J. L. Obesity, senescence, and senolytics. Handb. Exp. Pharmacol. 274, 165–180 (2022).
Montégut, L., Lopez-Otin, C., Magnan, C. & Kroemer, G. Old paradoxes and new opportunities for appetite control in obesity. Trends Endocrinol. Metab. 32, 264–294 (2021).
Sharma, A. M. & Engeli, S. Obesity and the renin–angiotensin–aldosterone system. Expert Rev. Endocrinol. Metab. 1, 255–264 (2006).
Khafagy, R. & Dash, S. Obesity and cardiovascular disease: the emerging role of inflammation. Front. Cardiovasc. Med. 8, 768119 (2021).
Ryder, J. R. et al. Accelerated early vascular aging among adolescents with obesity and/or type 2 diabetes mellitus. J. Am. Heart Assoc. 9, e014891 (2020).
Niemann, B. et al. Obesity induces signs of premature cardiac aging in younger patients: the role of mitochondria. J. Am. Coll. Cardiol. 57, 577–585 (2011).
de Cabo, R. & Mattson, M. P. Effects of intermittent fasting on health, aging, and disease. N. Engl. J. Med. 381, 2541–2551 (2019).
Meydani, S. N. et al. Long-term moderate calorie restriction inhibits inflammation without impairing cell-mediated immunity: a randomized controlled trial in non-obese humans. Aging 8, 1416–1431 (2016).
Spadaro, O. et al. Caloric restriction in humans reveals immunometabolic regulators of health span. Science 375, 671–677 (2022).
Stekovic, S. et al. Alternate day fasting improves physiological and molecular markers of aging in healthy, non-obese humans. Cell Metab. 31, 878–881 (2020).
Sabatine, M. S. et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 376, 1713–1722 (2017).
Bangalore, S., Kumar, S., Wetterslev, J. & Messerli, F. H. Angiotensin receptor blockers and risk of myocardial infarction: meta-analyses and trial sequential analyses of 147 020 patients from randomised trials. Br. Med. J. 342, d2234 (2011).
Granger, C. B. et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet 362, 772–776 (2003).
Cohn, J. N., Tognoni, G.; Valsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N. Engl. J. Med. 345, 1667–1675 (2001).
Pfeffer, M. A. et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet 362, 759–766 (2003).
McMurray, J. J. V. et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 371, 993–1004 (2014).
Flather, M. D. et al. Long-term ACE-inhibitor therapy in patients with heart failure or left-ventricular dysfunction: a systematic overview of data from individual patients. ACE-Inhibitor Myocardial Infarction Collaborative Group. Lancet 355, 1575–1581 (2000).
Dagenais, G. R., Pogue, J., Fox, K., Simoons, M. L. & Yusuf, S. Angiotensin-converting-enzyme inhibitors in stable vascular disease without left ventricular systolic dysfunction or heart failure: a combined analysis of three trials. Lancet 368, 581–588 (2006).
van Vark, L. C. et al. Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin-angiotensin-aldosterone system inhibitors involving 158,998 patients. Eur. Heart J. 33, 2088–2097 (2012).
SOLVD Investigators et al. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N. Engl. J. Med. 325, 293–302 (1991).
CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N. Engl. J. Med. 316, 1429–1435 (1987).
Packer, M. et al. Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 106, 2194–2199 (2002).
Flather, M. D. et al. Randomized trial to determine the effect of nebivolol on mortality and cardiovascular hospital admission in elderly patients with heart failure (SENIORS). Eur. Heart J. 26, 215–225 (2005).
Pitt, B. et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 341, 709–717 (1999).
Zannad, F. et al. Eplerenone in patients with systolic heart failure and mild symptoms. N. Engl. J. Med. 364, 11–21 (2011).
Pitt, B. et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 348, 1309–1321 (2003).
Tabula Muris Consortium. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 583, 590–595 (2020).
Pinto, A. R. et al. Revisiting cardiac cellular composition. Circ. Res. 118, 400–409 (2016).
Acknowledgements
The authors thank the reviewers for their valuable constructive feedback and apologize to the authors of several high-quality articles on cardiovascular ageing that could not be discussed and cited owing to space limitations. The authors are also grateful to Leen Assil Companioni (CBmed, Graz, Austria) for designing the figures before submission, Sylvère Durand (Institute Gustave Roussy, Villejuif, France) for spermidine measurements as well as Nathaly Anto Michel (Medical University of Graz, Graz, Austria) and Andrew L. Koenig (Washington University School of Medicine, St. Louis, MO, USA) for extracting and providing the single-cell data reported in Fig. 1. M.A. acknowledges support from the European Commission (H2020-MSCA-IF), the Austrian Society of Cardiology (Präsidentenstipendium-ÖKG), the Medical University of Graz (Start Fund), BioTechMed-Graz (Young Researcher Group) and the Austrian Science Fund (FWF; P34926). P.P.R. and S.S. acknowledge support from the Medical University of Graz (Flagship project VASC-HEALTH) and BioTechMed-Graz (Flagship project INTERACD+). G.K. is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; European Research Council Advanced Investigator Grand ‘ICD-Cancer’, Fondation pour la Recherche Médicale (FRM); a donation by Elior; Equipex Onco-Pheno-Screen; European Joint Programme on Rare Diseases (EJPRD); European Research Council (ICD-Cancer), European Union Horizon 2020 Projects Oncobiome and Crimson; Fondation Carrefour; Institut National du Cancer (INCa); Institut Universitaire de France; LabEx Immuno-Oncology (ANR-18-IDEX-0001); a Cancer Research ASPIRE Award from the Mark Foundation; the RHU Immunolife; Seerave Foundation; SIRIC Stratified Oncology Cell DNA Repair and Tumour Immune Elimination (SOCRATE); and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001.
Ethics declarations
Competing interests
M.A. and S.S. are involved in patent applications related to the cardiometabolic effects of caloric restriction mimetics. G.K. holds research contracts with Daiichi Sankyo, Eleor, Kaleido, Lytix Pharma, Osasuna Therapeutics, PharmaMar, Samsara Therapeutics, Sanofi, Tollys and Vascage. G.K. is a consultant for Reithera and is on the Board of Directors of the Bristol Myers Squibb Foundation, France. G.K. is also a scientific co-founder of everImmune, Osasuna Therapeutics, Samsara Therapeutics and Therafast Bio. G.K. is the inventor of patents covering therapeutic targeting of ageing, cancer, cystic fibrosis and metabolic disorders. G.K.’s wife, Laurence Zitvogel, has held research contracts with 9 Meters Biopharma, Daiichi Sankyo and Pilege, was on the Board of Directors of Transgene, is a co-founder of everImmune, and holds patents covering the treatment of cancer and therapeutic manipulation of the microbiota. G.K.’s brother, Romano Kroemer, was an employee of Sanofi and consulted for Boehringer-Ingelheim. P.P.R. declares no competing interests.
Peer review
Peer review information
Nature Reviews Cardiology thanks Luigi Fontana and the other, anonymous, reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Autophagy
-
An evolutionarily conserved homeostatic process through which old, dysfunctional or potentially hazardous cytoplasmic components undergo lysosomal degradation; three types of autophagy exist in mammalian cells: microautophagy, macroautophagy and chaperone-mediated autophagy.
- Clonal haematopoiesis of indeterminate potential
-
(CHIP). A process by which stem cells in the blood or bone marrow randomly accrues somatic mutations during ageing, leading to exaggerated representation of a single clone.
- Inflammageing
-
The phenomenon of low-grade, subclinical sterile inflammation commonly observed with ageing, leading to increased circulating and tissue cytokine levels.
- Inflammasome
-
A supramolecular complex, the formation of which is triggered by a range of exogenous and endogenous pro-inflammatory factors, culminating in the activation of caspase 1, which proteolytically activates the pro-inflammatory cytokines IL-1β and IL-18.
- Non-coding RNAs
-
(ncRNAs). RNA molecules that are not translated into proteins but regulate the expression levels of target genes; there are several subfamilies of ncRNAs, including microRNAs (<200 nucleotides), long non-coding RNAs (>200 nucleotides) and covalently closed circular RNAs.
- Proteostasis
-
A network of cellular processes that maintain the health and integrity of the proteome, including protein biogenesis, folding, trafficking and degradation.
- Senescence
-
Permanent arrest of the cell cycle with irreversible loss of replicative capacity, leading to the loss of specific cellular functions, and the acquisition of a secretory and metabolically active phenotype with pro-inflammatory features.
- Senolytic therapy
-
Elimination of senescent cells by molecules that specifically induce cell death in senescent cells to improve organismal health.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Abdellatif, M., Rainer, P.P., Sedej, S. et al. Hallmarks of cardiovascular ageing. Nat Rev Cardiol 20, 754–777 (2023). https://doi.org/10.1038/s41569-023-00881-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41569-023-00881-3
Subjects
This article is cited by
-
Caloric restriction and its mimetics in heart failure with preserved ejection fraction: mechanisms and therapeutic potential
Cardiovascular Diabetology (2025)
-
Roles and mechanisms of histone methylation in vascular aging and related diseases
Clinical Epigenetics (2025)
-
Biological age prediction and NAFLD risk assessment: a machine learning model based on a multicenter population in Nanchang, Jiangxi, China
BMC Gastroenterology (2025)
-
Genetic targets related to aging for the treatment of coronary artery disease
BMC Medical Genomics (2025)
-
Mitochondrial dysfunction in the regulation of aging and aging-related diseases
Cell Communication and Signaling (2025)